Protein structure is the bridge between genome and function — and the ability to predict, analyse, and exploit three-dimensional protein structures computationally has been fundamentally transformed by AlphaFold2 and the broader revolution in AI-powered structural biology. From structure prediction and binding site identification to molecular docking, virtual screening, molecular dynamics simulation, and protein-protein interaction analysis, structural bioinformatics now sits at the centre of rational drug design, protein engineering, and mechanistic biological research. At BioinformaticsNext, we provide specialist AlphaFold and structural bioinformatics services — supporting pharmaceutical, biotech, and academic programmes with expert computational structural biology from target characterisation through to structure-guided lead optimisation.
AlphaFold & Structural Bioinformatics: AI-Powered Protein Structure for Drug Discovery & Research
From AlphaFold2 structure prediction and binding site analysis to molecular docking, virtual screening, and molecular dynamics — enabling structure-guided drug discovery without the need for experimental crystallography.
The release of AlphaFold2 by DeepMind — and the subsequent expansion of the AlphaFold Protein Structure Database to cover virtually the entire known proteome — has democratised access to high-confidence 3D protein structures for targets that previously lacked any structural information. Combined with advances in molecular docking, molecular dynamics simulation, and AI-powered virtual screening, computational structural biology can now guide drug discovery programmes from the earliest stages of target identification through to lead optimisation and selectivity profiling.
At BioinformaticsNext, we provide the full structural bioinformatics stack — integrating AlphaFold2 structure prediction, experimental structure analysis, binding site characterisation, molecular docking, virtual screening, MD simulation, and protein engineering support to accelerate structure-guided drug discovery and protein research programmes with speed, accuracy, and scientific rigour.
What We Support
Comprehensive computational structural biology across protein structure prediction, drug-target interaction analysis, and protein engineering applications.
- AlphaFold2, RoseTTAFold, and ESMFold protein structure prediction for novel and understudied targets
- Protein complex and multimer structure prediction for protein-protein interaction analysis
- Binding site identification, druggability assessment, and allosteric pocket detection
- Molecular docking of small molecules, peptides, and fragments against target structures
- Large-scale virtual screening of compound libraries for hit identification and ranking
- Molecular dynamics (MD) simulations for protein-ligand complex stability and binding free energy
- Pharmacophore modelling and 3D shape-based virtual screening
- Protein-protein interaction (PPI) interface analysis and hot-spot residue identification
- Protein engineering support: stability prediction, mutation effect scoring, and directed evolution guidance
- Comparative structure analysis, homology modelling, and structural alignment
Our AlphaFold & Structural Bioinformatics Services
Expert computational structural biology services — from AI-powered structure prediction through to drug-target interaction analysis and protein engineering support.
All analyses are tailored to your target protein, therapeutic modality, compound series, and stage of discovery — from early target characterisation through to IND-enabling structural biology packages.
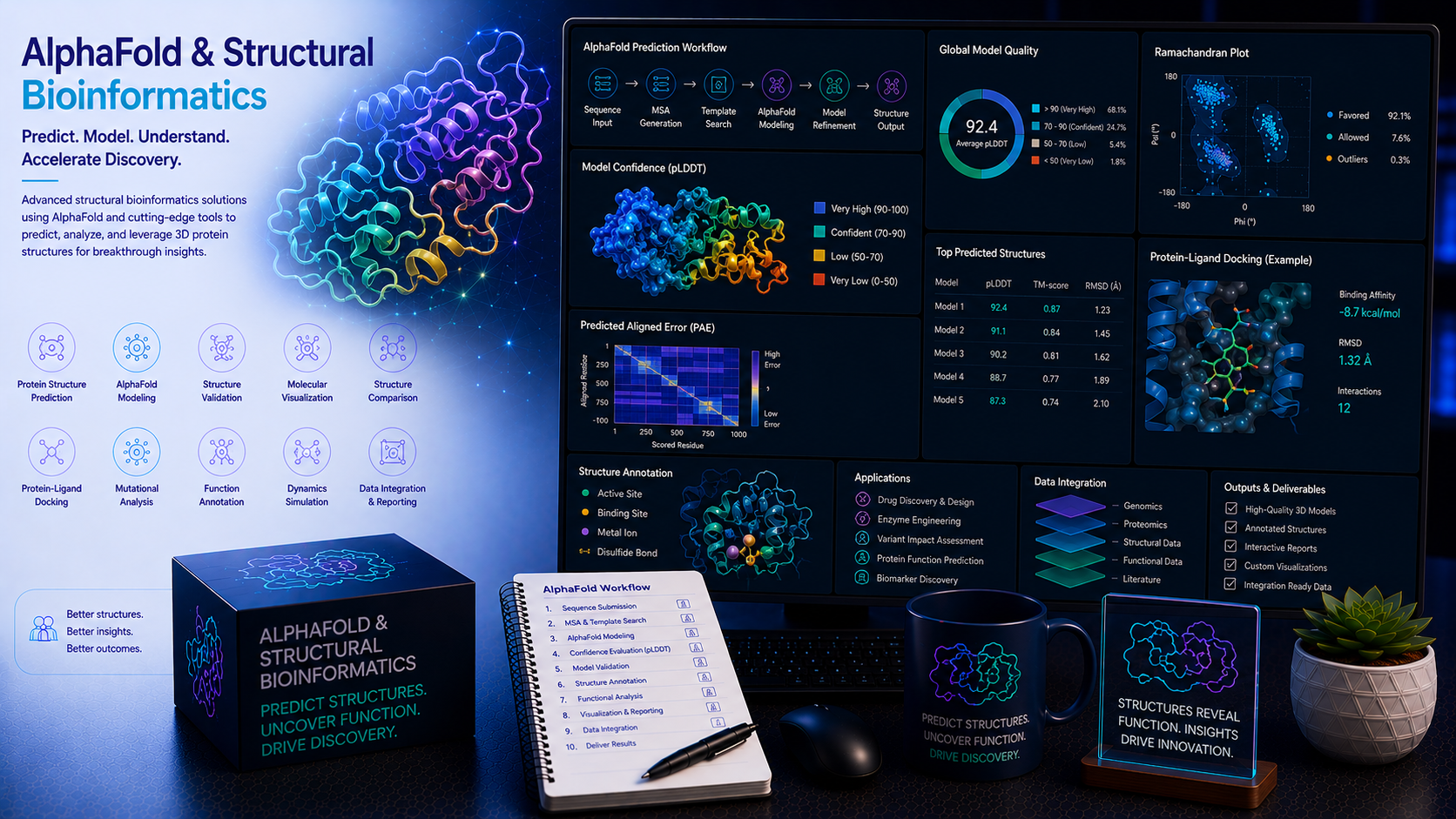
1. AlphaFold2 & AI Protein Structure Prediction AlphaFold2 · RoseTTAFold · ESMFold · Multimer
AlphaFold2 has redefined what is computationally achievable in protein structure prediction — delivering near-crystallographic accuracy for many protein families without the need for experimental structure determination. We deploy AlphaFold2, RoseTTAFold, ESMFold, and related models to generate, validate, and interpret high-confidence 3D protein structures for drug discovery and research applications.
- AlphaFold2 monomer structure prediction — Full AlphaFold2 pipeline execution with multiple sequence alignment (MSA) generation; per-residue pLDDT confidence scoring; predicted aligned error (PAE) map analysis for domain orientation confidence; structure quality validation with MolProbity and PROCHECK
- Protein complex and multimer prediction — AlphaFold-Multimer prediction of homo- and heterodimeric complexes; protein-protein interface geometry assessment; comparison with experimental PDB structures for benchmarking; antibody-antigen and enzyme-substrate complex modelling
- RoseTTAFold and ESMFold complementary prediction — Independent structure prediction with RoseTTAFold2 and ESMFold for model cross-validation; ensemble model generation for capturing conformational uncertainty in flexible regions
- Intrinsically disordered region (IDR) analysis — IUPred3, PONDR, and flDPnn disorder prediction; identification of ordered structural domains within predominantly disordered proteins; context-dependent folding prediction for disorder-to-order transitions
- Model confidence assessment and quality filtering — Systematic pLDDT and PAE threshold-based filtering of high- and low-confidence structural regions; identification of regions requiring experimental validation or alternative modelling strategies
2. Binding Site Identification & Druggability Assessment fpocket · SiteMap · DoGSiteScorer · Allosteric
Identifying tractable binding sites on a target protein — including orthosteric active sites, allosteric pockets, and cryptic sites that emerge only in certain conformational states — is the essential first step in structure-guided drug discovery. We apply multiple complementary computational approaches to comprehensively map the druggable surface of your target.
- Binding pocket detection and characterisation — fpocket, SiteMap, DoGSiteScorer, and P2Rank-based cavity detection on AlphaFold2-predicted and experimental structures; pocket volume, depth, hydrophobicity, and aromaticity scoring; druggability score calculation and site ranking
- Allosteric site identification — AlloSigMA, CryptoSite, and MDpocket-based allosteric pocket detection; molecular dynamics-based cryptic site identification from conformational ensemble analysis; allosteric communication pathway mapping with Bio3D and ProDy
- Active site comparison and conservation analysis — Binding site structural superposition against homologous proteins; selectivity pocket identification; conservation scoring of binding site residues across species and paralogues for selectivity window assessment
- Fragment hotspot mapping — FTMap and Fpocket fragment hotspot analysis; identification of high-affinity sub-pockets within binding sites for fragment-based drug design and fragment-to-lead optimisation guidance
3. Molecular Docking & Virtual Screening AutoDock Vina · Glide · GOLD · ZINC · ChEMBL
Molecular docking predicts the preferred binding pose and interaction energy of a small molecule within a protein binding site — enabling the rapid prioritisation of compound libraries for experimental testing. Combined with large-scale virtual screening, docking accelerates hit identification and reduces the cost and timelines of early drug discovery.
- Rigid and flexible molecular docking — AutoDock Vina, GOLD, and Glide-based small molecule docking against defined binding sites; induced-fit docking (IFD) for flexible receptor accommodating large or unusual scaffolds; docking score analysis, pose clustering, and interaction diagram generation
- Fragment docking — Docking of fragment libraries (Maybridge, Enamine FBDD) against target binding sites; fragment hotspot correlation; fragment growing and merging trajectory design for hit-to-lead progression
- Large-scale virtual screening — Screening of ChEMBL, ZINC20, Enamine REAL, and custom compound libraries (up to tens of millions of compounds) against target structures; multi-stage screening funnels combining pharmacophore pre-filtering, docking, and ADMET filters for hit ranking
- Covalent docking — CovDock and AutoDock-based covalent docking for targeted covalent inhibitor design; warhead reactivity and selectivity assessment; cysteine, lysine, and serine residue covalent binding site identification
- Peptide and macrocycle docking — FlexPepDock and RosettaDock-based peptide docking against protein binding sites; macrocycle conformational sampling and docking for PROTAC linker design and cyclic peptide programmes
4. Molecular Dynamics Simulations & Binding Free Energy GROMACS · AMBER · MM-PBSA · FEP
Molecular docking provides a static snapshot of protein-ligand interaction — but protein-ligand binding is a dynamic process. Molecular dynamics (MD) simulations capture the conformational flexibility of both the protein and the ligand over time, enabling assessment of binding pose stability, binding free energy estimation, and identification of cryptic binding sites unavailable from static crystal structures.
- Protein and protein-ligand MD simulations — GROMACS and AMBER force field-based MD simulations of apo proteins and protein-ligand complexes; CHARMM36 and ff14SB force field parameterisation; GAFF2 and CGenFF ligand parameterisation; trajectory analysis including RMSD, RMSF, radius of gyration, and contact map evolution
- Binding free energy estimation — MM-PBSA and MM-GBSA end-point binding free energy calculation; per-residue energy decomposition for hotspot residue identification; relative binding free energy ranking across congeneric compound series
- Free energy perturbation (FEP) calculations — Alchemical FEP and thermodynamic integration for high-accuracy relative binding free energy prediction; RBFE calculations for lead optimisation compound ranking within a scaffold series
- Enhanced sampling and metadynamics — Replica exchange MD (REMD) and metadynamics simulations for conformational landscape exploration; cryptic binding site detection from conformational ensemble analysis; protein-ligand unbinding pathway characterisation
- Membrane protein simulations — GPCR, ion channel, and transporter MD simulations in explicit lipid bilayer environments; CHARMM-GUI membrane builder setup; lipid composition effects on protein conformation and drug binding
5. Protein-Protein Interaction (PPI) Analysis & Interface Druggability Hot-Spots · PPI Inhibitors · PROTAC · Glue
Protein-protein interactions (PPIs) regulate virtually every cellular process and represent an increasingly important — though challenging — class of drug targets. We provide specialist computational PPI analysis to characterise interaction interfaces, identify hot-spot residues, and assess tractability for small molecule, peptide, or molecular glue disruption or stabilisation.
- PPI interface characterisation — AlphaFold-Multimer and HADDOCK-based protein complex structure prediction; buried surface area, interface residue identification, and shape complementarity analysis; comparison with experimental PDB complex structures
- Hot-spot residue prediction — FoldX, Robetta, and computational alanine scanning mutagenesis for binding hot-spot residue identification; energetic contribution ranking; validation against available mutagenesis data
- PPI interface druggability assessment — fpocket and SiteMap-based interface pocket detection; hot-spot clustering for small molecule binding site identification; assessment of interface tractability for inhibitor, stabiliser, or molecular glue approaches
- PROTAC and molecular glue target assessment — Ternary complex modelling of PROTAC-mediated target-E3 ligase complexes with AlphaFold-Multimer and ClusPro; linker length and geometry optimisation; molecular glue-induced neo-interface characterisation
- Antibody and biologics epitope mapping — Structural epitope prediction and linear epitope mapping; antibody-antigen complex modelling with AlphaFold-Multimer and HDock; paratope residue identification and antibody binding mode characterisation
6. Pharmacophore Modelling & SBDD 3D Pharmacophore · Shape Screening · Scaffold Hopping
Pharmacophore models capture the essential three-dimensional arrangement of chemical features required for potent binding to a target — enabling scaffold-independent virtual screening, hit expansion, and the identification of structurally diverse active series when existing leads are constrained by IP, ADMET, or selectivity limitations.
- Structure-based 3D pharmacophore generation — Pharmit, Phase, and MOE-based pharmacophore model generation from protein-ligand complex structures and docking poses; feature selection, refinement, and validation against known actives and inactives
- Ligand-based pharmacophore modelling — Common feature pharmacophore generation from aligned active compound sets; activity-annotated pharmacophore model development; pharmacophore-based VS for novel scaffold identification
- 3D shape and electrostatic screening — ROCS and Pharmit shape-based screening of large compound collections; electrostatic overlay scoring; scaffold hopping to structurally diverse actives maintaining target pharmacophore geometry
- Structure-based drug design (SBDD) support — Iterative docking, pharmacophore, and free energy-guided design cycle support; medicinal chemistry hypothesis generation from structural analysis; SAR rationalisation from docking pose visualisation
7. Protein Engineering & Stability Analysis FoldX · EvolutionaryScale · Thermostability · Design
Computational protein engineering enables rational design of proteins with improved thermostability, solubility, binding affinity, and manufacturability — reducing experimental screening burden and accelerating the development of enzymes, antibodies, therapeutic proteins, and biosensors. We provide AI-assisted and physics-based protein engineering support across academic and industrial applications.
- Mutation effect prediction — FoldX, Rosetta ddG, and DynaMut2-based prediction of the thermodynamic effect of single and multiple mutations on protein stability (ΔΔG); identification of stabilising mutations for therapeutic protein and biosensor engineering
- Evolutionary sequence analysis for engineering — Multiple sequence alignment-based conservation analysis; EVmutation and ESM-1v evolutionary coupling scores for mutation tolerance prediction; identification of positions tolerant to substitution without structural disruption
- AI-guided protein design — ProteinMPNN and RFdiffusion-based sequence design for novel protein structures; EvolutionaryScale ESM3-guided sequence optimisation for stability and function; de novo protein binder design for target epitopes
- Antibody and nanobody engineering — CDR loop modelling and humanisation assessment; affinity maturation candidate identification from structural analysis; bispecific antibody format geometry evaluation and linker design
- Enzyme active site engineering — Rosetta enzyme design and QM/MM-guided catalytic residue optimisation; substrate specificity and enantioselectivity prediction; directed evolution library design informed by structural analysis
Key Applications
Structural bioinformatics applications across drug discovery, protein research, and biotechnology development.
- Structure prediction for novel and understudied drug targets lacking experimental structures
- Binding site and druggability assessment for target tractability evaluation
- Virtual screening for hit identification against ChEMBL, ZINC, and Enamine libraries
- Structure-guided lead optimisation and selectivity profiling
- PPI interface analysis for molecular glue, PROTAC, and inhibitor design
- Fragment-based drug discovery computational support
- MD simulations for binding pose validation and binding free energy ranking
- Cryptic and allosteric binding site identification for difficult-to-drug targets
- Antibody and nanobody structural characterisation and engineering
- Enzyme engineering for industrial biotechnology and biosensor development
- Protein stability and solubility optimisation for biologic manufacturing
- Structural biology support for IND and regulatory submission packages
Our Analytical Workflow
A structured, reproducible structural bioinformatics process tailored to your target, compound series, and stage of drug discovery.
Step 1 — Project Scoping Free
We discuss your target protein, available structural data, compound series, therapeutic modality, and discovery objectives to define the most appropriate structural bioinformatics approach and deliverables — at no cost.
Step 2 — Structure Acquisition & Prediction
PDB experimental structure retrieval and assessment; AlphaFold2 or RoseTTAFold structure prediction for targets lacking experimental data; homology model generation where appropriate; structure quality validation.
Step 3 — Binding Site Analysis
Multi-tool binding pocket detection and characterisation; druggability scoring; allosteric site identification; active site conservation and selectivity analysis; fragment hotspot mapping.
Step 4 — Docking & Virtual Screening
Compound library preparation and filtering; multi-stage virtual screening funnel; molecular docking and pose scoring; pharmacophore and shape-based screening; ADMET pre-filtering; ranked hit list generation.
Step 5 — MD Simulation & Free Energy Analysis
System preparation, force field parameterisation, and MD simulation execution; trajectory analysis and binding pose stability assessment; MM-PBSA/MM-GBSA or FEP binding free energy calculation for compound ranking.
Step 6 — Structural Analysis & Engineering
PPI interface hot-spot analysis; mutation effect prediction; pharmacophore model generation; protein engineering candidate identification; antibody or enzyme structural assessment as appropriate to your programme.
Step 7 — Visualisation & Reporting
Publication-ready figures — PyMOL and ChimeraX structure visualisations, docking pose interaction diagrams, binding site surface maps, MD trajectory plots, and free energy landscapes — with a full written scientific report.
Step 8 — Regulatory & IP Support Optional
Structural biology sections for IND and regulatory submissions; patent application computational structural biology content; manuscript preparation and supplementary figure legends; ongoing structure-guided lead optimisation retainer support.
Tools & Technologies
Industry-standard and cutting-edge structural bioinformatics tools across all protein structure, docking, simulation, and engineering workflows.
- Structure Prediction: AlphaFold2, AlphaFold-Multimer, RoseTTAFold2, ESMFold, ColabFold, OpenFold
- Homology Modelling: MODELLER, Swiss-Model, Phyre2, I-TASSER
- Structure QC: MolProbity, PROCHECK, WHATCHECK, PDBeFold
- Binding Site Detection: fpocket, SiteMap, DoGSiteScorer, P2Rank, FTMap, MDpocket
- Molecular Docking: AutoDock Vina, AutoDock-GPU, GOLD, Glide, PLANTS, FlexPepDock
- Covalent & Peptide Docking: CovDock, RosettaDock, HADDOCK, HDock
- MD Simulations: GROMACS, AMBER, NAMD, OpenMM, CHARMM-GUI
- Free Energy: gmx_MMPBSA, FEP+, Yank, TIES, alchemical-analysis
- Pharmacophore & VS: Pharmit, Phase, ROCS, LigandScout, MOE
- Protein Engineering: FoldX, Rosetta Suite, DynaMut2, ProteinMPNN, RFdiffusion, ESM3
- Visualisation: PyMOL, UCSF ChimeraX, VMD, NGLview, ProDy, Bio3D
- Cheminformatics: RDKit, Open Babel, Schrodinger LigPrep, ADMET-AI, pkCSM
Reference Databases We Use
All major structural, chemical, and biological databases supporting protein structure analysis, virtual screening, and drug discovery.
- Protein Data Bank (PDB) — Repository of experimentally determined protein, nucleic acid, and complex structures; primary source for template selection, docking validation, and binding site benchmarking
- AlphaFold Protein Structure Database — DeepMind and EMBL-EBI AlphaFold2 structure predictions covering the human proteome and key model organism proteomes; first-pass structural reference for novel targets
- UniProt / UniRef — Protein sequence and functional annotation database; MSA source for AlphaFold2 prediction and evolutionary analysis; domain boundary and post-translational modification reference
- ChEMBL — Bioactivity database of drug-like compounds and drug targets; compound library for virtual screening; target-activity data for docking validation and QSAR model benchmarking
- ZINC20 — Large-scale purchasable compound library of over 750 million compounds for virtual screening campaigns; pre-filtered in-stock and make-on-demand compound subsets
- Enamine REAL / REAL Space — Synthetically accessible compound library of over 6 billion compounds for ultra-large virtual screening; docking-ready 3D compound libraries
- BindingDB & PDBbind — Curated protein-ligand binding affinity databases for docking validation, QSAR modelling, and binding free energy method benchmarking
- COSMIC Cancer Gene Census — Oncogene and tumour suppressor annotations for structural characterisation of cancer-associated mutations and hotspot residue identification
Project Deliverables
A complete, structured set of structural bioinformatics outputs to advance your drug discovery or protein research programme.
- AlphaFold2 predicted structures in PDB format with pLDDT confidence maps and PAE plots
- Binding site report: pocket coordinates, druggability scores, volume, and residue lining analysis
- Docking results: ranked compound poses, docking scores, interaction fingerprints, and 2D/3D interaction diagrams
- Virtual screening hit list with scores, ADMET predictions, and purchasability information
- MD simulation analysis: RMSD/RMSF plots, binding pose stability assessment, and trajectory summary
- Binding free energy estimates with confidence intervals and compound ranking
- Publication-ready structure visualisations (PyMOL/ChimeraX scenes, PNG/SVG at 300 dpi)
- Full written scientific report: methods, results, interpretation, and structure-guided recommendations
- Pipeline scripts and configuration files for complete analytical reproducibility
- IND and regulatory submission structural biology sections
- Patent application computational structural biology content
- Manuscript methods section and supplementary figure legends (journal-formatted)
- Interactive 3D structure viewer for internal team use
- Ongoing structure-guided lead optimisation retainer support
- Grant application computational structural biology sections and preliminary data
- FEP calculations for high-accuracy compound ranking within a lead series
Why Choose BioinformaticsNext?
Expert computational structural biology — combining cutting-edge AI structure prediction with rigorous molecular modelling to accelerate your drug discovery programme.
AlphaFold & AI Structure Expertise
We go beyond running AlphaFold2 — we critically assess model confidence, identify regions requiring experimental validation, and integrate predicted structures into downstream docking, MD, and engineering workflows with appropriate rigour.
Drug Discovery Context
Our structural bioinformatics is always framed in its drug discovery context — every binding site analysis, docking campaign, and MD simulation is interpreted in terms of chemical tractability, selectivity, ADMET implications, and medicinal chemistry actionability.
End-to-End Structural Biology Stack
From AlphaFold2 structure prediction and binding site mapping through virtual screening, MD simulation, free energy calculation, and protein engineering — we cover the complete computational structural biology pipeline in a single engagement.
Difficult Target Expertise
We have specialist experience with challenging target classes — intrinsically disordered proteins, protein-protein interactions, membrane proteins, allosteric targets, and covalent drug targets — where standard docking approaches require careful adaptation.
Fast Turnaround
Most structural bioinformatics projects are delivered within 2–4 weeks. Virtual screening campaigns and MD simulations may require additional time depending on library size and simulation length — discussed and agreed at project scoping.
IP & Data Security
Strict NDAs, encrypted data transfer, and IP protection protocols as standard. All compound structures, target sequences, and structural data are treated as strictly confidential and never shared beyond the agreed project scope.
Flexible Engagement
Project-based, milestone-driven, or long-term retainer arrangements for ongoing structure-guided lead optimisation. We integrate seamlessly with your medicinal chemistry, computational chemistry, and drug discovery teams.
Global Reach
UK-headquartered with clients across Europe, North America, the Middle East, and Asia-Pacific. Full remote collaboration with encrypted data transfer and secure compound library handling as standard.
Frequently Asked Questions
Common questions from pharmaceutical, biotech, and academic clients about AlphaFold and structural bioinformatics services.
AlphaFold2 structures are highly reliable for well-conserved globular protein domains — often matching or exceeding the resolution of low-to-medium resolution crystal structures. However, reliability varies by region: high pLDDT scores (>90) indicate high confidence, while low-scoring regions (pLDDT <70) should be treated with caution. We critically assess every AlphaFold2 model, identify low-confidence regions, and advise on which structural conclusions can be drawn reliably — and where experimental validation should be prioritised.
Yes. We use AlphaFold-Multimer for homo- and heterodimeric complex prediction, including protein-protein, protein-peptide, and antibody-antigen complexes. We validate complex models against experimental data where available, assess interface confidence using PAE maps, and compare predicted interfaces with known homologous complexes in the PDB.
We screen against all major publicly available compound libraries — ChEMBL, ZINC20, Enamine REAL Space (up to billions of compounds), Maybridge, and LifeChemicals — as well as proprietary compound libraries provided by the client. Multi-stage screening funnels incorporating pharmacophore pre-filtering, docking, ADMET prediction, and diversity analysis are used to deliver a manageable, high-quality hit list for experimental follow-up.
Molecular docking provides a rapid, computationally economical estimate of binding pose and affinity — but treats the protein as largely rigid and does not capture binding dynamics. MD simulations are most valuable when: (1) you need to assess binding pose stability over time, (2) the protein is known to be flexible or undergoes conformational selection, (3) you need binding free energy estimates more accurate than docking scores, or (4) you are investigating an allosteric or cryptic binding site. We advise on the most appropriate level of computational treatment for each project at scoping.
Yes. We have experience modelling GPCRs, ion channels, transporters, and other membrane-spanning proteins using AlphaFold2-predicted structures embedded in explicit lipid bilayer environments with CHARMM-GUI. We perform binding site analysis, ligand docking, and MD simulations in physiologically relevant membrane environments — including the effects of lipid composition on receptor conformation and drug binding.
Yes. We provide ternary complex modelling of PROTAC-mediated target–E3 ligase complexes using AlphaFold-Multimer and protein-protein docking; linker length, geometry, and flexibility assessment; and cooperative binding energy estimation. For molecular glues, we characterise glue-induced neo-interface contacts and assess interface druggability of the induced complex.
Absolutely. We assist with the computational structural biology sections of grant applications — including AlphaFold2 structure prediction plans, virtual screening methodology, MD simulation approaches, and preliminary structural data. Please contact us as early as possible in the grant preparation process to allow time for any preliminary analyses required.
Related Research Areas & Services
AlphaFold and structural bioinformatics connects to and supports multiple complementary research areas and services we provide.
- Drug Development & AI-Driven Discovery — QSAR modelling, ADMET prediction, generative molecular design, drug-target interaction prediction, and drug repurposing — all informed by and integrated with structural analysis
- AI Drug Target Identification — Druggability assessment of AI-nominated targets using AlphaFold2 structural analysis; binding site tractability scoring as part of multi-evidence target prioritisation
- Genetics & Genomics — Structural interpretation of GWAS-prioritised missense variants; AlphaFold2-based protein structural impact assessment of genetic variants identified in disease cohorts
- Cancer & Oncogenomics — Structural characterisation of cancer driver mutations and oncogenic hotspot residues; structural basis for drug resistance mutation analysis in precision oncology
- Cell & Gene Therapy Bioinformatics — Structural analysis of AAV capsid variants and engineered capsid proteins; CRISPR-Cas structural modelling for guide RNA and PAM specificity optimisation
- Custom Software & Pipeline Development — Bespoke structural bioinformatics pipelines, automated docking workflows, and interactive structure-activity relationship (SAR) visualisation platforms for internal drug discovery teams
Ready to Advance Your Structure-Guided Drug Discovery Programme?
Tell us about your target protein, your compound series, and your discovery objectives. Our AlphaFold and structural bioinformatics team will design a tailored computational plan — typically within 48 hours of your enquiry. Whether you need structure prediction for an unstudied target, a virtual screening campaign against a new binding site, MD simulations to rank your lead compounds, or protein engineering support, we are here to accelerate your programme from day one.
