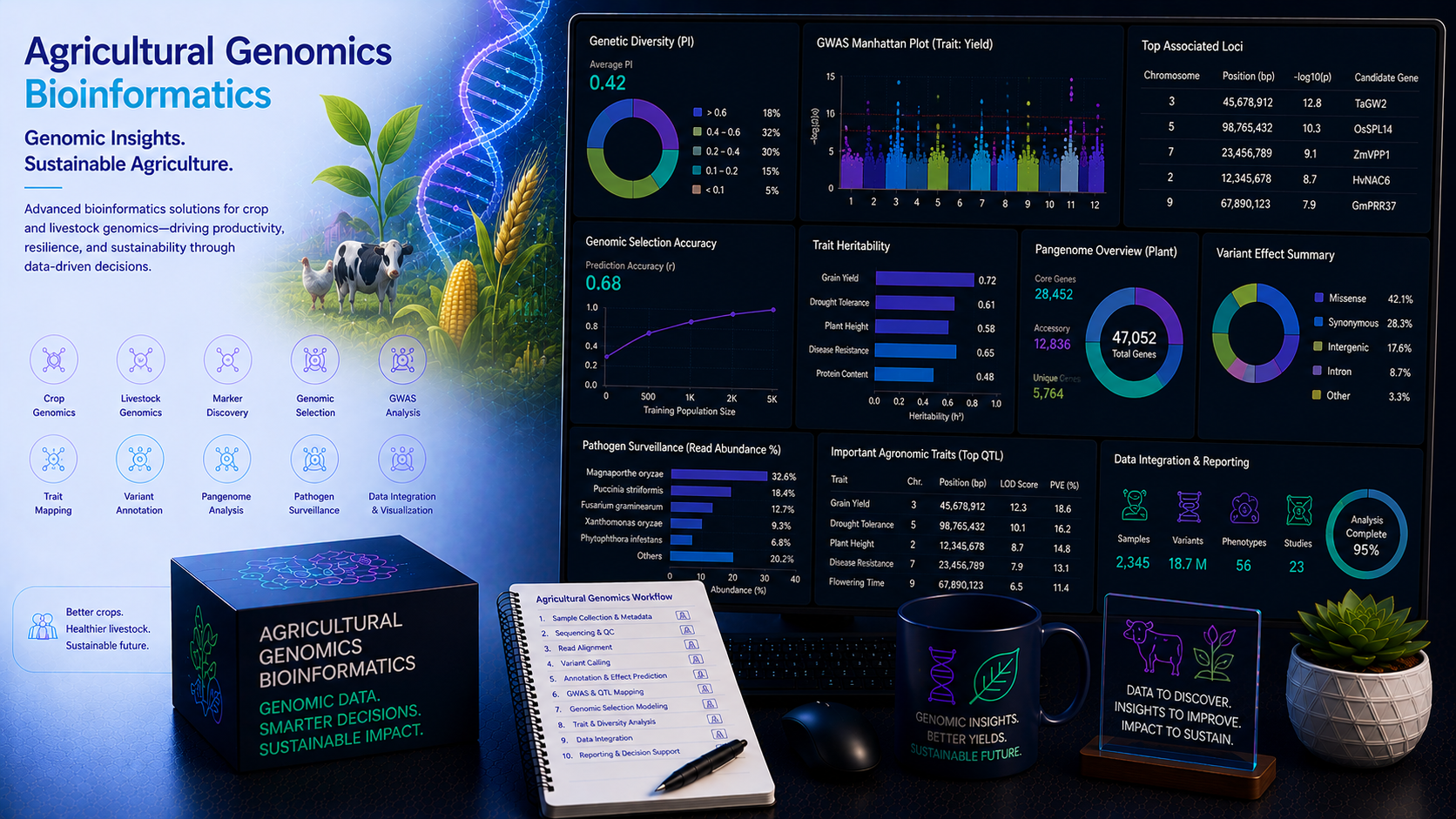
Agricultural genomics is transforming crop science and livestock breeding — accelerating the identification of genes and variants underlying agronomically important traits, enabling genomic selection for yield, disease resistance, drought tolerance, and nutritional quality, and revealing the full genomic diversity of crop species through pan-genome construction. From GWAS-based quantitative trait locus (QTL) mapping and marker-assisted selection to reference genome assembly, comparative genomics, and epigenomic regulation of agronomic traits, agricultural genomics generates complex, large-scale datasets that demand expert bioinformatics analysis. At BioinformaticsNext, we provide specialist agricultural genomics bioinformatics services — supporting plant breeders, crop science research institutions, agri-biotech companies, and livestock genomics programmes with rigorous, reproducible, and translatable computational analyses.
Agricultural Genomics Bioinformatics: Crop Genomics, Trait Selection & Pan-Genomes
Expert bioinformatics for crop and livestock GWAS, genomic selection, QTL mapping, pan-genome construction, comparative genomics, and epigenomic trait analysis — accelerating plant and animal breeding programmes with data-driven precision.
The global challenges of food security, climate change adaptation, and sustainable agriculture demand faster, more precise crop and livestock improvement than conventional phenotypic selection alone can deliver. Genomic tools now underpin every stage of modern plant breeding — from the discovery of causal genetic variants underlying complex agronomic traits through genomic estimated breeding value (GEBV) prediction and marker-assisted selection to pan-genome construction capturing the full gene content diversity of a crop species. The polyploid, highly repetitive genomes of major crop species such as wheat, cotton, and sugarcane, combined with the massive linkage disequilibrium in some domesticated populations, create unique bioinformatics challenges not encountered in human genomics. At BioinformaticsNext, we provide the full agricultural genomics bioinformatics stack — adapted to the specific genomic architectures, reference resources, and breeding objectives of major crop species and livestock programmes.
What We Support
Comprehensive agricultural genomics bioinformatics across crop and livestock GWAS, genomic selection, genome assembly, pan-genome construction, and epigenomic trait analysis.
- Crop and livestock GWAS for agronomic, disease resistance, and quality trait mapping
- QTL mapping, fine mapping, and candidate gene identification from bi-parental and multi-parent populations
- Genomic selection and genomic estimated breeding value (GEBV) prediction for accelerated breeding
- Reference genome assembly, annotation, and quality assessment for crop species
- Pan-genome construction and accessory genome characterisation for crop genetic diversity
- Comparative genomics and synteny analysis across crop species and wild relatives
- Population genomics, domestication sweep detection, and genetic diversity assessment
- Crop epigenomics: DNA methylation, chromatin accessibility, and histone modification profiling
- Transcriptomic analysis of stress response, developmental stages, and trait-associated gene expression
- Marker development, genotyping-by-sequencing (GBS), and SNP array design support
Our Agricultural Genomics Bioinformatics Services
Specialist crop and livestock genomics bioinformatics — from GWAS and QTL mapping through genomic selection, pan-genome construction, and epigenomic trait analysis.
All analyses are tailored to your crop or livestock species, genomic resources, breeding population structure, and research or commercial objectives.
1. Crop & Livestock GWAS and QTL Mapping GWAS · QTL · BLUP · MLM · Fine Mapping
Genome-wide association studies and QTL mapping identify the genomic regions and causal variants underlying agronomically important traits — from yield components and disease resistance to grain quality, drought tolerance, and animal production traits. We apply statistically rigorous, population structure-corrected association methods appropriate for the unique linkage disequilibrium, relatedness, and polyploidy characteristics of crop and livestock genomes.
- GWAS for agronomic traits — GAPIT3, GEMMA, and FarmCPU-based mixed linear model (MLM) GWAS with population structure (Q) and kinship (K) correction; compressed MLM, BLINK, and multi-locus MRMLM for improved power; Bonferroni and FDR-based significance thresholds; Manhattan and QQ plot generation with genomic inflation factor assessment
- QTL mapping in bi-parental and multi-parent populations — R/qtl and R/qtl2-based interval mapping and composite interval mapping (CIM) in F2, RIL, DH, and BC populations; multi-parent advanced generation intercross (MAGIC) and nested association mapping (NAM) population QTL analysis; QTL confidence interval estimation and epistasis testing
- Fine mapping and candidate gene identification — QTL fine mapping using near-isogenic line (NIL) and introgression line (IL) populations; haplotype block analysis and linkage disequilibrium mapping; candidate gene prioritisation from physical interval gene annotation, expression data, and functional evidence
- Polyploid-aware association analysis — Dosage-sensitive SNP calling and association testing in allopolyploid species (wheat, cotton, oilseed rape, sugarcane); polyploid GWAS with PolyGWAS and GWASpoly; homeologous chromosome discrimination and subgenome-specific association analysis
2. Genomic Selection & Breeding Value Prediction GEBV · GBLUP · rrBLUP · Machine Learning · GxE
Genomic selection uses genome-wide SNP marker data to predict the breeding value of individuals for complex quantitative traits — enabling selection decisions before phenotyping is complete and dramatically accelerating breeding cycles. We develop, validate, and optimise genomic selection models for yield, quality, resistance, and production traits in crops and livestock.
- Genomic estimated breeding value (GEBV) prediction — rrBLUP, GBLUP, and BayesA/B/C genomic prediction models; genomic relationship matrix (GRM) construction and inversion; cross-validation prediction accuracy assessment; training population optimisation for maximum genomic prediction accuracy
- Multi-trait and multi-environment genomic selection — Multi-trait GBLUP models for correlated agronomic traits; genotype-by-environment (GxE) interaction modelling with reaction norm and factor analytic models; across-environment GEBV stability analysis for target environment selection
- Machine learning genomic prediction models — Random forest, gradient boosting, deep learning (genomic CNN, LSTM), and reproducing kernel Hilbert spaces (RKHS) genomic prediction models; comparison of ML vs. GBLUP prediction accuracy across trait architectures; feature importance analysis for SNP effect estimation
- Genomic selection pipeline development — End-to-end genomic selection pipeline from raw genotype data to GEBV ranking for breeding decisions; marker QC, imputation, and GRM construction automation; periodic model retraining with updated phenotypic data integration
3. Reference Genome Assembly & Annotation HiFi · Hi-C · Scaffolding · Repeat Masking · Annotation
A high-quality reference genome is the foundation of all downstream agricultural genomics — enabling accurate read mapping, variant calling, gene model annotation, and comparative analysis. We provide end-to-end genome assembly and annotation for crop and wild relative species, with expertise in the large, repetitive, and polyploid genomes that are common in agriculturally important plant species.
- Long-read genome assembly — PacBio HiFi (hifiasm, HiCanu) and Oxford Nanopore (Flye, Shasta) assembly for chromosome-level crop genome assembly; hybrid assembly combining long-read contigs with Illumina short-read polishing; assembly quality assessment with BUSCO, QUAST, and Merqury
- Hi-C scaffolding and chromosome-level assembly — 3D-DNA, YAHS, and Salsa2-based Hi-C chromatin contact scaffolding to chromosome scale; pseudomolecule construction and centromere identification; comparison with genetic linkage map for scaffolding validation
- Repeat masking and transposable element annotation — RepeatModeler2 and RepeatMasker de novo repeat library construction and genome-wide TE annotation; LTR retrotransposon dating for evolutionary analysis; TE content comparison across related crop species and wild relatives
- Gene model annotation — MAKER2, BRAKER2, and EvidenceModeler-based structural annotation combining ab initio prediction, RNA-seq evidence, and homology; functional annotation with InterProScan, BLAST against UniProt/SwissProt, and GO term assignment; annotation quality assessment with BUSCO completeness scoring
4. Pan-Genome Construction & Comparative Genomics Pan-Genome · Core Genome · Accessory Genes · Synteny
No single reference genome captures the full genetic diversity of a crop species — particularly the presence/absence variation (PAV) of genes that underlies important agronomic trait differences between varieties. Pan-genome construction integrates multiple high-quality assemblies to characterise the core genome shared by all accessions and the accessory genome present in only a subset, revealing the full genic and structural diversity available for crop improvement.
- Pan-genome graph construction — Minigraph-Cactus, PGGB, and VG-based pangenome graph assembly from multiple crop accession assemblies; core, softcore, shell, and cloud genome partitioning; presence/absence variation (PAV) matrix construction across accessions
- Pan-genome analysis and functional characterisation — Core genome GO term enrichment for essential biological processes; accessory genome functional enrichment for defence, stress response, and secondary metabolism; pan-genome size estimation and gene accumulation curve modelling
- Synteny and comparative genomics — MCScan, SynChro, and MUMmer-based whole-genome synteny analysis between crop species and wild relatives; syntenic block identification and collinearity visualisation; large-scale structural variant and chromosomal rearrangement mapping between genomes
- Structural variant discovery from pan-genome — Large insertion, deletion, inversion, and translocation identification from multi-assembly comparison; SV-trait association analysis linking pan-genomic structural variation to agronomic phenotypes; SV genotyping across diverse germplasm collections
5. Crop Population Genomics, Domestication & Epigenomics Selective Sweeps · Diversity · Methylation · Stress Response
Understanding the population genomic history of crop species — the demographic bottlenecks of domestication, the selective sweeps marking breeding improvement, and the epigenomic regulation of developmental and stress-responsive gene expression — provides the evolutionary context for agronomic trait variation and identifies underexploited genetic diversity in crop wild relatives for introgression.
- Population structure and genetic diversity assessment — SNP-based PCA, ADMIXTURE, and TreeMix population structure analysis across diverse crop accessions and wild relatives; nucleotide diversity (π), Tajima's D, and FST differentiation statistics; genetic erosion assessment comparing modern varieties with landraces and wild progenitors
- Domestication sweep and selection signature detection — XP-EHH, iHS, and CLR-based selective sweep detection between wild and domesticated populations; FST outlier analysis for divergently selected genomic regions; candidate domestication gene identification from sweep regions overlapping annotated genes and QTL
- Crop epigenomics analysis — WGBS and RRBS DNA methylation profiling in CG, CHG, and CHH contexts across crop genomes; differentially methylated region (DMR) identification between developmental stages, stress conditions, and genotypes; MethylKit, DSS, and Bismark-based methylation analysis; chromatin accessibility (ATAC-seq) profiling in crop tissues
- Stress response and developmental transcriptomics — DESeq2 and edgeR differential expression analysis across drought, heat, salinity, and pathogen stress conditions; co-expression network construction with WGCNA for hub gene and module identification; time-course transcriptomic modelling of developmental stage transitions
Key Applications
Agricultural genomics bioinformatics across major crop species, livestock programmes, and plant breeding applications.
- Wheat, maize, rice, soybean, and barley yield and quality GWAS
- Disease resistance QTL mapping for rust, blight, and blast pathogens
- Genomic selection for hybrid performance and heterosis prediction
- Pan-genome construction for allopolyploid crop species
- Drought and heat stress tolerance gene discovery and marker development
- Livestock SNP array design and genomic estimated breeding value prediction
- Crop wild relative diversity assessment for introgression breeding
- Epigenomic regulation of seed quality and stress memory traits
Tools, Technologies & Reference Databases
Validated, widely adopted agricultural genomics bioinformatics tools and all major crop genomics reference resources.
- GWAS & QTL: GAPIT3, GEMMA, FarmCPU, BLINK, R/qtl2, GWASpoly, PolyGWAS
- Genomic Selection: rrBLUP, GBLUP (sommer), BayesA/B/C (BGLR), ASReml, AlphaImpute
- Genome Assembly: hifiasm, HiCanu, Flye, 3D-DNA, YAHS, Salsa2, BUSCO, Merqury
- Annotation: MAKER2, BRAKER2, EvidenceModeler, RepeatModeler2, InterProScan
- Pan-Genome: Minigraph-Cactus, PGGB, VG toolkit, MCScan, MUMmer, SyRI
- Population Genomics: PLINK, ADMIXTURE, TreeMix, SweepFinder2, XP-EHH (selscan)
- Epigenomics: Bismark, BSseq, methylKit, DSS, MACS2, HOMER, deepTools
- Transcriptomics: STAR, HISAT2, DESeq2, edgeR, WGCNA, clusterProfiler
- Ensembl Plants / Gramene — Crop genome assemblies, gene annotations, and comparative genomics resources
- URGI / Phytozome / MaizeGDB / IWGSC — Crop-specific genome portals for wheat, maize, and major cereals
Project Deliverables
Structured, publication-ready agricultural genomics bioinformatics outputs for every project.
- GWAS results: Manhattan plots, QQ plots, significant SNP tables, and candidate gene lists
- QTL mapping outputs: LOD score profiles, confidence intervals, and additive effect estimates
- GEBV prediction accuracy report with cross-validation results and model comparison
- Genome assembly statistics: N50, BUSCO completeness, and Merqury QV score
- Pan-genome core/accessory gene classification and PAV matrix
- Population structure plots: PCA, ADMIXTURE, and phylogenetic trees with diversity statistics
- Publication-ready figures (PDF/SVG/PNG at 300 dpi)
- Full written scientific report with methods, results, and breeding or research recommendations
- Pipeline scripts and configuration files for complete analytical reproducibility
- Marker-assisted selection (MAS) panel design from GWAS or QTL results
- Genomic selection pipeline development for internal breeding programme deployment
- Pan-genome graph browser and interactive synteny visualisation tools
- Crop wild relative diversity and introgression target identification report
- Manuscript methods section and supplementary figure legends
- Grant application agricultural genomics sections with preliminary data
- Long-term retainer for ongoing breeding programme genomic analysis support
Frequently Asked Questions
Common questions from plant breeders, crop science researchers, and agri-biotech companies.
Polyploid GWAS requires specialised approaches at every step — from SNP calling that accounts for allele dosage (rather than simple diploid genotypes) to association testing models that handle the inflated false positive rates from homeologous chromosome cross-mapping. We use GWASpoly and PolyGWAS for dosage-based association testing, apply subgenome-specific alignment strategies to discriminate homeologous loci in allopolyploids, and use mixed linear models with polyploid-appropriate kinship matrices to control population structure. We have specific experience with hexaploid wheat (IWGSC reference), tetraploid cotton, and autotetraploid potato genomic analyses.
Genomic prediction accuracy depends on training population size, marker density, trait heritability, and LD between markers and causal variants. As a general guide, training populations of 500–2,000 lines with 5,000–50,000 SNPs provide reasonable genomic prediction accuracy for moderately heritable traits in most self-pollinating crop species. For cross-pollinating species and livestock with lower LD, larger training populations and higher marker densities are required. We provide power calculations and training population optimisation analysis at project scoping, and can evaluate your existing dataset for prediction accuracy before committing to a full genomic selection programme.
A pan-genome captures the complete gene content of a species — comprising the core genome (genes present in all accessions), the softcore genome (genes in most accessions), and the accessory genome (genes present in only some accessions). In crop species, the accessory genome can contain 20–40% of total gene content and often includes genes for disease resistance, stress tolerance, and secondary metabolism that are absent from any single reference genome. Pan-genome construction from multiple diverse accession assemblies enables accurate genotyping of presence/absence variation (PAV) across breeding germplasm and reveals the full functional diversity available for crop improvement — including genes present in wild relatives but absent from elite varieties.
Yes. We provide de novo genome assembly and annotation for crop species without existing reference genomes — using PacBio HiFi or Oxford Nanopore long-read sequencing combined with Hi-C chromatin contact data for chromosome-level scaffolding. Our assembly workflow includes genome size estimation (flow cytometry or k-mer analysis), long-read assembly, Hi-C scaffolding, repeat masking, and structural and functional gene annotation. We assess assembly quality with BUSCO completeness, Merqury QV score, and comparison with any available genetic linkage maps before delivering the final annotated assembly.
Absolutely. We assist with the bioinformatics and computational genomics sections of agricultural genomics grant applications — including proposed GWAS and QTL workflows, genomic selection methodology, pan-genome construction approaches, and preliminary genomic data. We have experience supporting applications to BBSRC, UKRI, Innovate UK, EU Horizon, and USDA-NIFA funding programmes. Please contact us as early as possible in the grant preparation process to allow time for any preliminary analyses.
Related Research Areas & Services
Agricultural genomics bioinformatics connects to multiple complementary services we support.
- Genetics & Genomics — Population genetics, GWAS methodology, demographic history inference, and variant analysis approaches shared between agricultural and human genomics applications
- eDNA & Biodiversity Genomics — Soil and rhizosphere microbial community profiling, plant-microbe interaction genomics, and crop wild relative diversity assessment from environmental samples
- Metagenomics & Microbiome Analysis — Soil microbiome and plant endophyte community profiling relevant to crop health, rhizobiome optimisation, and sustainable agriculture research
- Structural & Functional Genomics — Chromatin accessibility, histone modification, and regulatory element analysis in crop development, stress response, and epigenetic trait inheritance
- AI & Machine Learning Applications — Machine learning genomic prediction models, deep learning-based trait prediction, and knowledge graph approaches adapted from drug discovery to crop trait prioritisation
- Custom Software & Pipeline Development — Bespoke agricultural genomics analysis platforms, automated GEBV reporting pipelines, and genomic selection workflow deployment for plant breeding programmes
Ready to Advance Your Agricultural Genomics Programme?
Tell us about your crop or livestock species, your genomic data, your breeding objectives, and your research or commercial goals. Our agricultural genomics bioinformatics team will design a tailored analytical plan — typically within 48 hours of your enquiry. Whether you need polyploid GWAS and QTL mapping, genomic selection model development, pan-genome construction, reference genome assembly and annotation, or population genomics for crop improvement, we are here to deliver expert, reproducible agricultural genomics results from day one.
