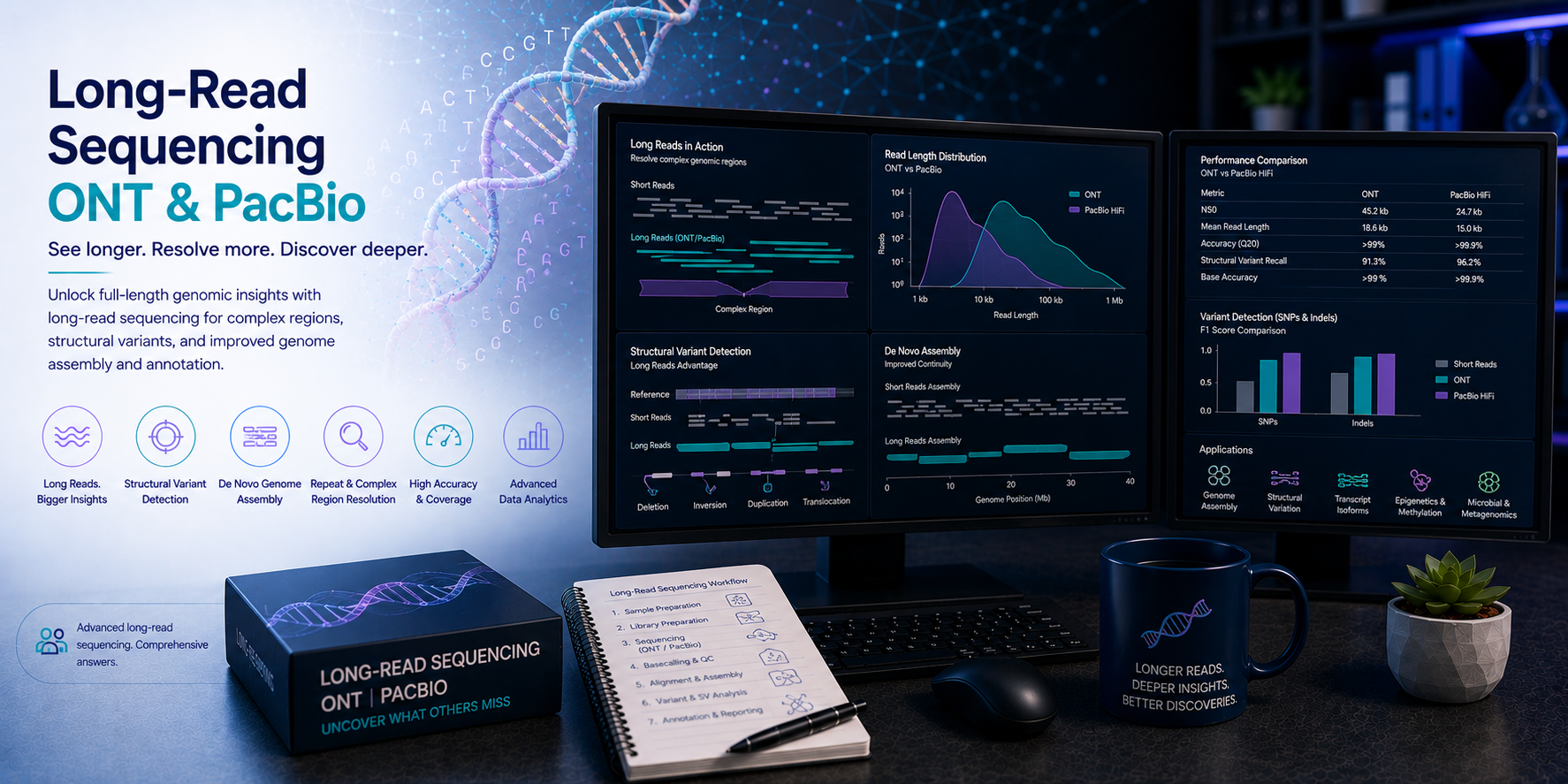
Long-read sequencing technologies — Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio) — have overcome the fundamental limitation of short-read sequencing by generating reads spanning thousands to hundreds of thousands of base pairs. This read length transforms what is computationally achievable: complex structural variants invisible to Illumina sequencing are resolved with single-base precision; full-length mRNA isoforms are sequenced end-to-end without assembly; repetitive and highly homologous genomic regions are spanned and phased; complete microbial genomes are assembled from a single sequencing run; and methylation is detected directly from the raw electrical signal without bisulphite conversion. At BioinformaticsNext, we provide specialist long-read sequencing bioinformatics — supporting research groups, clinical genomics laboratories, pharmaceutical companies, and genome centres in extracting the full analytical value from ONT and PacBio long-read data across all biological applications.
Long-Read Sequencing Bioinformatics: ONT & PacBio for Structural Variants, Isoforms & Beyond
Expert bioinformatics for Oxford Nanopore and PacBio long-read sequencing data — including structural variant detection, full-length isoform characterisation, genome assembly, complex locus phasing, direct epigenetic modification detection, and clinical long-read genomics applications.
The human genome contains millions of structural variants — deletions, duplications, inversions, translocations, and mobile element insertions — that collectively account for more base pairs of variation between individuals than all single nucleotide variants combined. Yet the vast majority of these variants are invisible to short-read sequencing because their breakpoints lie within repetitive sequences that reads of 150 bp cannot span. Similarly, the transcriptome contains thousands of alternative isoforms generated by complex combinations of alternative splicing, alternative promoter usage, and alternative polyadenylation — but short-read RNA-seq cannot sequence full-length transcripts and must instead infer isoform structure through computational assembly, introducing substantial uncertainty. Long-read sequencing resolves both challenges directly — and unlocks additional capabilities in genome assembly, epigenomics, and clinical genomics that are simply not accessible with short reads alone.
What We Support
Comprehensive long-read sequencing bioinformatics across all major ONT and PacBio platforms, data types, and biological applications.
- Structural variant (SV) detection and genotyping from ONT and PacBio WGS data
- Full-length isoform sequencing: ONT cDNA/direct RNA and PacBio Iso-Seq analysis
- De novo and reference-guided long-read genome assembly and annotation
- Complex locus phasing: HLA, CYP2D6, SMN1/2, and highly repetitive regions
- Direct epigenetic modification detection: 5mC, 5hmC, and 6mA from ONT signal
- Repeat expansion detection and characterisation (STR, VNTR, satellite repeats)
- Long-read metagenomics for complete microbial genome recovery
- Adaptive sampling and targeted enrichment with Oxford Nanopore
- Clinical long-read genomics: rare disease, cancer structural genomics, and WGS reporting
- Hybrid assembly combining long-read contigs with short-read polishing
Our Long-Read Sequencing Bioinformatics Services
Specialist ONT and PacBio bioinformatics — from basecalling and QC through structural variant analysis, isoform characterisation, genome assembly, epigenomics, and clinical long-read reporting.
All analyses are tailored to your sequencing platform, library preparation chemistry, coverage depth, target application, and biological or clinical research objectives.
1. Long-Read Data Processing, QC & Alignment Dorado · Guppy · Minimap2 · NanoStat · HiFi · PBMM2
Rigorous basecalling, quality assessment, and alignment are the essential foundation of every long-read analysis — with platform-specific considerations for ONT signal processing, PacBio CCS consensus accuracy, and the unique alignment challenges of reads spanning repetitive and structurally complex genomic regions.
- ONT basecalling and QC — Dorado and Guppy-based basecalling with appropriate model selection (fast, hac, sup) for each flow cell chemistry (R9.4.1, R10.4.1); duplex basecalling for R10 data; NanoStat, NanoPlot, and PycoQC read length distribution, quality score, and yield QC; per-barcode demultiplexing for multiplexed runs
- PacBio HiFi CCS generation and QC — SMRT Link and ccs-based circular consensus sequence (CCS) read generation from raw subreads; zmw pass filtering and minimum accuracy thresholds; HiFi read length, quality, and yield QC; lima-based multiplexed sample demultiplexing and adapter trimming
- Long-read alignment — Minimap2 splice-aware alignment for RNA and genome mapping; PBMM2 alignment of PacBio HiFi reads to reference genomes; LRA and Winnowmap for highly repetitive region alignment; secondary and supplementary alignment filtering; coverage uniformity and depth reporting across target regions
- Targeted long-read sequencing QC — Adaptive sampling (ReadFish, BOSS-RUNS) run monitoring and target enrichment efficiency assessment; targeted panel coverage analysis; on-target rate, enrichment fold, and uniformity reporting for amplicon and capture-based long-read approaches
2. Structural Variant Detection & Genotyping Sniffles2 · PBSV · DELLY · SVision · SV Genotyping
Structural variants — deletions, duplications, inversions, translocations, insertions, and mobile element insertions — are the most impactful class of genetic variation by base pairs affected, yet the most poorly characterised due to the limitations of short-read sequencing. Long-read SV detection resolves complex rearrangements at single-base-pair breakpoint precision and enables SV genotyping across population cohorts.
- Comprehensive SV detection — Sniffles2 and PBSV-based SV calling from ONT and PacBio alignments; DELLY and SVABA complementary short-read SV integration for hybrid SV calling; SV type classification: deletions, duplications, inversions, translocations, insertions, and complex rearrangements; SURVIVOR SV merging and consensus calling across multiple callers
- Mobile element insertion (MEI) characterisation — TLDR, PALMER, and Sniffles2 MEI mode-based LINE-1, Alu, SVA, and HERV insertion detection; insertion site characterisation and target site duplication analysis; MEI allele frequency estimation across population samples; somatic MEI detection in cancer genomes
- Complex SV and chromosomal rearrangement analysis — SVision and PBSV complex SV classification; chromothripsis and chromoplexy detection from cancer long-read WGS; breakpoint junction sequence characterisation; templated insertion chain identification; copy number profiling from long-read coverage data
- Population-scale SV genotyping — Paragraph, Bayestyper, and SVABA-based SV genotyping in additional short-read samples from long-read-derived SV callsets; population allele frequency estimation; SV-GWAS for trait association; structural variant phasing with haplotype-resolved long-read data
3. Full-Length Isoform Sequencing & Transcriptome Characterisation Iso-Seq · FLAMES · StringTie2 · IsoQuant · Novel Isoforms
Full-length isoform sequencing with PacBio Iso-Seq and ONT cDNA or direct RNA sequencing eliminates the isoform assembly uncertainty of short-read RNA-seq — sequencing each transcript end-to-end to provide definitive characterisation of the full splice site combination, TSS, and poly-A site of each expressed isoform. This resolves isoform diversity in complex loci, identifies disease-associated novel isoforms, and enables accurate isoform-level differential expression analysis.
- PacBio Iso-Seq processing and isoform calling — SMRT Link Iso-Seq3 and isoseq3 CLI-based CCS generation, primer removal, poly-A tail trimming, and full-length non-concatemer (FLNC) read clustering; minimap2 genome alignment; SQANTI3 isoform structural annotation and quality classification; TAMA merge for multi-sample isoform catalogue construction
- ONT cDNA and direct RNA isoform analysis — FLAMES and IsoQuant-based isoform identification from ONT long reads; StringTie2 long-read mode transcript assembly; FLAIR isoform detection with short-read splice junction support for improved accuracy; direct RNA sequencing analysis preserving native RNA modifications
- Novel isoform identification and annotation — SQANTI3 structural classification of isoforms as FSM, ISM, NIC, NNC, antisense, intergenic, and genic; novel junction and novel splice site identification; NMD-susceptible isoform prediction; tissue-specific and condition-specific isoform expression comparison
- Isoform-level differential expression and splicing analysis — DEXSeq and DRIMseq isoform-level differential usage analysis; SUPPA2 alternative splicing event quantification from long-read isoform data; allele-specific isoform expression; poly-A site and TSS usage comparison across conditions; functional annotation of differentially expressed isoforms
4. Complex Locus Phasing, Repeat Expansions & Direct Epigenetics HLA · CYP2D6 · STR · 5mC · Modkit · Phasing
Some of the most clinically and biologically important genomic regions — HLA, CYP2D6, SMN1/2, repeat expansion loci, centromeres — are effectively inaccessible to short-read sequencing due to their complexity, length, or repetitiveness. Long-read sequencing resolves these regions definitively, and additionally detects DNA methylation directly from the electrical signal without any chemical conversion.
- Complex locus phasing and haplotype resolution — WhatsHap and HAPCUT2 long-read phasing; HLA typing with HLA-HD and T1K from long-read data to full eight-digit resolution; CYP2D6 star allele calling with Aldy and Cyrius from long-read data resolving duplication and hybrid alleles; SMN1/SMN2 copy number and sequence variant determination with LongReadSum and Paraphase
- Repeat expansion detection and characterisation — TRGT, Straglr, and DeepRepeat-based short tandem repeat (STR) expansion genotyping from long-read data; pathogenic repeat expansion confirmation in Huntington's (HTT CAG), fragile X (FMR1 CGG), C9orf72 (GGGGCC), myotonic dystrophy (DMPK CTG), and Friedreich's ataxia (FXN GAA); repeat interruption and motif variation analysis
- Direct methylation detection from ONT signal — Dorado and Modkit-based 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) detection from R10.4.1 ONT data without bisulphite conversion; per-read and per-CpG methylation probability calling; allele-specific methylation from phased reads; imprinting analysis and epigenetic clock estimation from long-read methylation data
- Centromere and satellite repeat analysis — T2T-CHM13-guided centromere assembly and analysis; alpha-satellite repeat monomer characterisation; CENP-A binding site identification from long reads; satellite repeat variant genotyping; centromeric structural variant and epiallele characterisation
5. Clinical Long-Read Genomics & Genome Assembly Rare Disease · Cancer · hifiasm · Chromosome-Scale · T2T
Long-read sequencing is increasingly entering clinical practice — resolving previously undiagnosed rare disease cases through SV detection and repeat expansion characterisation, enabling tumour structural genomics beyond what short-read panels can provide, and powering the assembly of high-quality reference genomes for model and non-model organisms. We provide specialist bioinformatics for clinical and research genome assembly and clinical long-read WGS reporting.
- Rare disease clinical long-read WGS analysis — Comprehensive SV, repeat expansion, and SNV calling from clinical long-read WGS; integration with short-read variant calls for maximum diagnostic yield; ACMG/AMP classification of structural and repeat expansion variants; trio-based phasing for de novo SV and compound heterozygous variant identification; previously unsolved case re-analysis with long-read data
- Cancer structural genomics — Tumour long-read WGS for comprehensive somatic SV calling including complex rearrangements, chromothripsis, and extrachromosomal DNA (ecDNA) detection; somatic MEI identification; structural variant-driven gene fusion identification; tumour suppressor disruption by SV; allele-specific copy number and LOH from phased long-read data
- De novo genome assembly — hifiasm, HiCanu, and Verkko PacBio HiFi assembly; Flye and Shasta ONT assembly; hybrid assembly combining ONT ultra-long reads with HiFi accuracy; Hi-C scaffolding with YAHS and 3D-DNA to chromosome scale; BUSCO, Merqury QV, and assembly statistics quality assessment; T2T-complete assembly strategies for small genomes
- Genome annotation and comparative genomics — MAKER2, BRAKER3, and EvidenceModeler structural annotation with long-read RNA evidence; RepeatModeler2 TE annotation; functional GO and InterPro annotation; SyRI and MUMmer whole-genome synteny and structural variant comparison between assemblies; pan-genome graph construction from multiple long-read assemblies
Key Applications
Long-read sequencing bioinformatics across clinical genomics, cancer research, transcriptomics, and genome science.
- Rare disease diagnosis from SV and repeat expansion long-read WGS
- Cancer tumour structural genomics and complex rearrangement characterisation
- Full-length isoform characterisation for disease transcriptome research
- HLA, CYP2D6, and SMN1/2 complex locus phasing for clinical genomics
- Direct 5mC and 5hmC methylation profiling without bisulphite conversion
- Chromosome-scale de novo genome assembly for model and non-model species
- Complete microbial genome recovery from long-read metagenomics
- Adaptive sampling and real-time targeted enrichment for clinical applications
Tools, Technologies & Reference Resources
Validated, cutting-edge long-read bioinformatics tools across all ONT and PacBio platforms and analysis workflows.
- Basecalling & QC: Dorado, Guppy, ccs (SMRT Link), NanoStat, NanoPlot, PycoQC
- Alignment: Minimap2, PBMM2, LRA, Winnowmap, Samtools, NGMLR
- SV Detection: Sniffles2, PBSV, DELLY, SVision, SURVIVOR, TLDR, PALMER
- SV Genotyping: Paragraph, Bayestyper, SVABA, Truvari, JASMINE
- Isoforms: Iso-Seq3, FLAMES, IsoQuant, StringTie2, FLAIR, SQANTI3, TAMA
- Phasing & Complex Loci: WhatsHap, HAPCUT2, HLA-HD, T1K, Aldy, Cyrius, Paraphase
- Repeat Expansions: TRGT, Straglr, DeepRepeat, ExpansionHunter Denovo
- Methylation: Modkit, Dorado (mod basecalling), f5c, MethPhase, Modbam2bed
- Genome Assembly: hifiasm, Verkko, Flye, HiCanu, YAHS, 3D-DNA, BUSCO, Merqury
- Annotation: MAKER2, BRAKER3, RepeatModeler2, InterProScan, SyRI, MUMmer
Project Deliverables
Structured, publication-ready long-read sequencing bioinformatics outputs for every project.
- Read QC report: length distribution, quality scores, yield, and per-barcode summary
- Alignment statistics: coverage depth, uniformity, and on-target rate
- SV callset in VCF format with type classification, size, genotype, and breakpoint sequences
- Isoform catalogue with SQANTI3 structural classification and expression quantification
- Repeat expansion genotype calls with confidence intervals and pathogenicity assessment
- Per-CpG methylation calls in bedMethyl format with allele-specific methylation (where applicable)
- Genome assembly statistics: N50, BUSCO completeness, Merqury QV, and chromosome-scale scaffolding metrics
- Publication-ready figures (PDF/SVG/PNG at 300 dpi)
- Full written scientific report with methods, results, interpretation, and recommendations
- Clinical rare disease ACMG/AMP SV and repeat expansion variant classification report
- Cancer somatic structural genomics report with gene fusion and ecDNA identification
- HLA, CYP2D6, and SMN1/2 clinical haplotype resolution report
- Genome annotation and comparative genomics synteny analysis
- Adaptive sampling pipeline development and real-time analysis configuration
- Manuscript methods section and supplementary figure legends
- Grant application long-read sequencing sections and preliminary data
- Long-term retainer for ongoing long-read sequencing programme support
Frequently Asked Questions
Common questions from clinical genomics laboratories, cancer research groups, and genome scientists.
PacBio HiFi (CCS) produces reads of 10–25 kb at very high accuracy (Q30, >99.9%) — making it the preferred choice for clinical variant calling, SNV and indel detection, phasing, and genome assembly where base-level accuracy is critical. Oxford Nanopore offers greater read length flexibility — from standard 10–50 kb reads to ultra-long reads of 100 kb–4 Mb with R10 chemistry — and additionally provides direct methylation detection from the raw signal without additional library preparation. ONT is preferred when ultra-long reads are needed to span very large structural variants or repetitive regions, for real-time adaptive sampling, and for direct RNA sequencing. For genome assembly, a hybrid approach using HiFi for accuracy and ONT ultra-long reads for spanning centromeres and large repeats provides the highest quality. We advise on platform selection at project scoping based on your specific application and budget.
Long-read SV detection has substantially higher sensitivity and precision than short-read approaches — particularly for insertions, inversions, and variants in repetitive regions. Short-read SV callers typically miss 50–70% of SVs detectable by long reads, particularly insertions larger than 50 bp and SVs with breakpoints in repetitive sequences. Long reads span SV breakpoints directly, enabling single-base-pair breakpoint resolution and precise characterisation of inserted sequences rather than inferring SVs from split read and discordant pair signals. For clinical applications where SV detection directly influences diagnosis — rare disease structural rearrangements, repeat expansions, and complex cancer structural genomics — long-read WGS provides dramatically improved diagnostic yield over short-read sequencing alone.
Yes — direct methylation detection is one of the most significant advantages of Oxford Nanopore sequencing. The R10.4.1 flow cell chemistry combined with Dorado's modification-aware basecalling model detects 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) directly from the current signal, providing genome-wide methylation calls at per-CpG resolution from a single WGS experiment without any additional library preparation. This eliminates bisulphite conversion-induced DNA degradation, enables methylation phasing relative to haplotype, and produces both variant calls and methylation profiles from a single sequencing run — substantially reducing cost and sample requirements compared to separate WGS and WGBS experiments.
Yes. Full-length isoform sequencing is well-suited to clinical and disease tissue samples — including FFPE material using ONT's low-input direct cDNA protocols for degraded RNA, fresh-frozen tissue with standard Iso-Seq or cDNA library preparation, and single-cell long-read isoform sequencing (MAS-Seq, FLAMES) for cell-type-resolved isoform characterisation. We have experience with isoform analysis in neurodegenerative disease, cancer, rare genetic disorders, and normal tissue atlases — identifying disease-associated novel splice isoforms, NMD-triggering transcripts, and allele-specific isoform expression patterns from long-read transcriptomics data.
Absolutely. We assist with the long-read sequencing bioinformatics sections of grant applications — including platform selection justification, SV analysis workflows, isoform characterisation methodology, genome assembly approaches, methylation analysis pipelines, and preliminary long-read data. Please contact us as early as possible in the grant preparation process to allow time for any preliminary analyses that would strengthen the scientific case for funding.
Related Research Areas & Services
Long-read sequencing bioinformatics connects to multiple complementary services we support.
- Clinical Genomics & Variant Interpretation — ACMG/AMP variant classification, rare disease trio analysis, and germline variant interpretation integrating long-read SV and repeat expansion findings into clinical diagnostic reports
- Cancer & Oncogenomics — Somatic variant calling, tumour mutational profiling, and structural genomics integration with long-read cancer WGS for comprehensive tumour characterisation
- Cell & Gene Therapy Bioinformatics — Long-read vector genome characterisation, ITR integrity assessment, integration site analysis, and CRISPR large structural variant detection from long-read sequencing data
- Pharmacogenomics (PGx) — Long-read phasing of complex PGx loci including CYP2D6 hybrid alleles, duplications, and deletions that are unresolvable by short-read star allele calling
- Agricultural Genomics — Long-read de novo crop genome assembly, pan-genome construction, and structural variant discovery for plant and livestock genomics programmes
- Custom Software & Pipeline Development — Bespoke long-read sequencing analysis platforms, automated SV reporting pipelines, adaptive sampling workflow development, and clinical long-read WGS reporting system deployment
Ready to Unlock the Full Power of Your Long-Read Sequencing Data?
Tell us about your ONT or PacBio platform, your library preparation chemistry, your target application, and your biological or clinical objectives. Our long-read sequencing bioinformatics team will design a tailored analytical plan — typically within 48 hours of your enquiry. Whether you need comprehensive SV detection and phasing, full-length isoform characterisation, chromosome-scale genome assembly, direct methylation profiling, complex locus resolution, or clinical long-read WGS reporting, we are here to deliver expert, publication-ready long-read results from day one.
