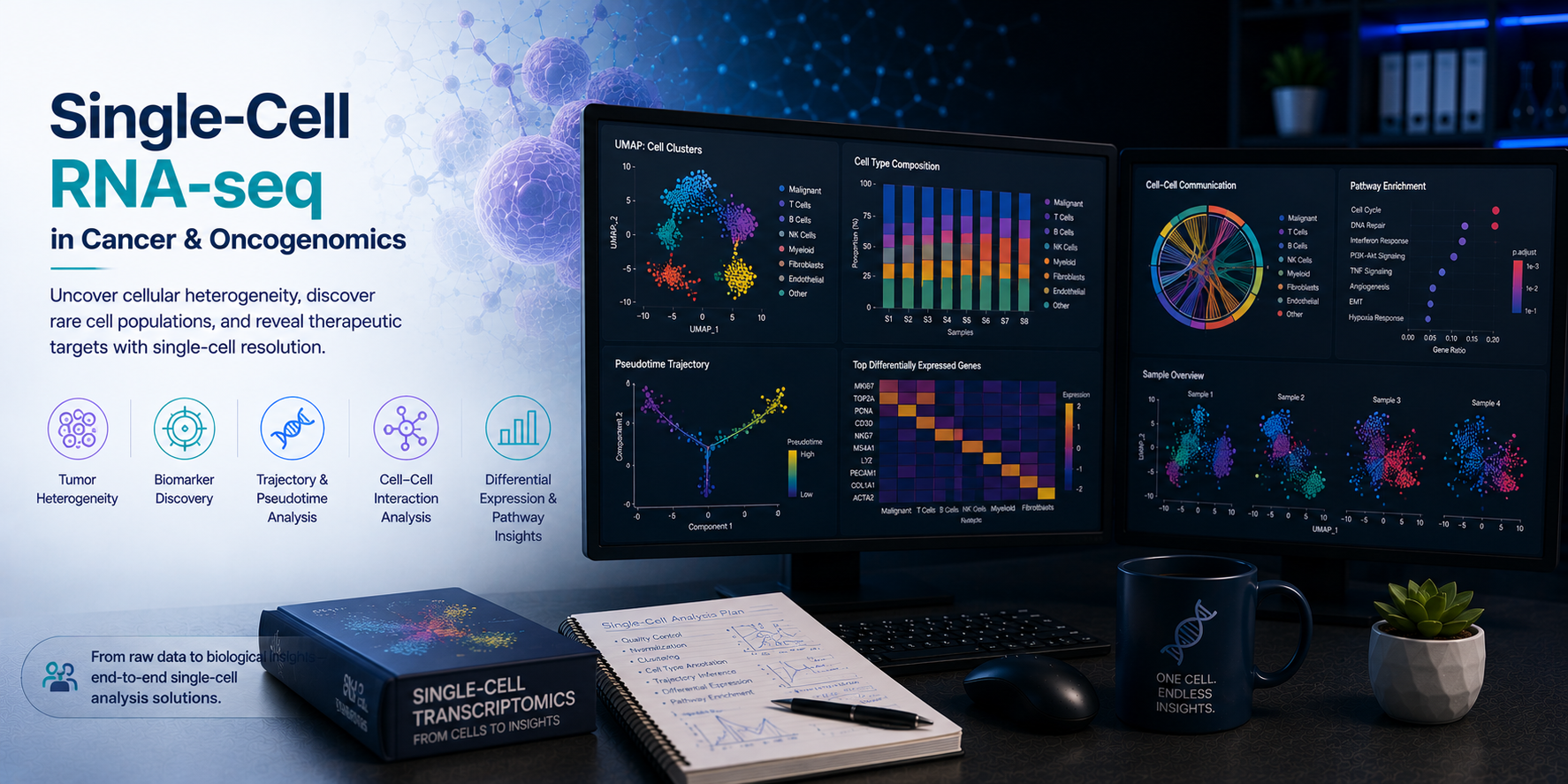
Single-cell RNA sequencing (scRNA-seq) has fundamentally transformed our understanding of tumour biology — resolving the cellular heterogeneity of tumours, immune infiltrates, and stromal populations at single-cell resolution, and enabling the study of clonal evolution, therapy resistance, and immune evasion with unprecedented depth. From tumour microenvironment (TME) immune cell deconvolution and cancer stem cell identification to clonal dynamics tracking and therapy-induced transcriptional reprogramming, scRNA-seq generates insights that bulk RNA-seq simply cannot provide. At BioinformaticsNext, we provide specialist scRNA-seq bioinformatics services focused on oncology — delivering expert tumour microenvironment profiling, clonal evolution analysis, and single-cell multi-omics integration for cancer research, drug discovery, and translational medicine programmes.
Single-Cell RNA-seq: Tumour Microenvironment & Clonal Evolution Bioinformatics
Expert scRNA-seq analysis for TME immune profiling, cancer cell state characterisation, clonal evolution tracking, therapy resistance, and single-cell multi-omics integration in oncology.
Tumours are not homogeneous masses — they are complex ecosystems comprising malignant cells in diverse transcriptional states, infiltrating immune populations spanning cytotoxic T cells, exhausted CD8+ T cells, immunosuppressive regulatory T cells, tumour-associated macrophages, NK cells, B cells, and dendritic cells, alongside cancer-associated fibroblasts, endothelial cells, and pericytes that shape the physical and immunological landscape of the tumour. Understanding how these populations interact, how malignant cells evolve under therapeutic pressure, and which cell states drive resistance or response to therapy is now directly accessible through scRNA-seq — but only with rigorous, expert bioinformatics analysis. At BioinformaticsNext, we provide the full scRNA-seq analytical stack for oncology — from raw data processing and cell type annotation to advanced TME profiling, clonal evolution reconstruction, and spatially-resolved tumour biology.
What We Support
Comprehensive single-cell RNA-seq bioinformatics for tumour microenvironment characterisation, clonal dynamics, therapy response, and multi-omics integration.
- scRNA-seq data processing, QC, and cell type annotation from all major platforms
- Tumour microenvironment immune cell deconvolution and functional state characterisation
- Cancer cell transcriptional state, stemness, and epithelial-mesenchymal transition (EMT) scoring
- Clonal evolution and copy number inference from scRNA-seq data
- Therapy resistance and drug response transcriptional programme identification
- Single-cell trajectory and pseudotime analysis of tumour and immune cell differentiation
- CITE-seq, 10x Multiome, and single-cell multi-omics integration
- Spatial transcriptomics of tumour tissue for TME spatial architecture mapping
- Cell-cell communication and ligand-receptor interaction analysis in the TME
- Integration of scRNA-seq with bulk RNA-seq, WGS, and clinical outcome data
Our scRNA-seq Oncology Bioinformatics Services
Specialist single-cell RNA-seq analysis for tumour biology, immune oncology, clonal evolution, and translational cancer research.
All analyses are tailored to your tumour type, sequencing platform, experimental design, sample source, and research or clinical objectives.
1. scRNA-seq Data Processing, QC & Cell Type Annotation Cell Ranger · Seurat · Scanpy · Harmony · Doublets
Rigorous data processing and accurate cell type annotation are the foundation of every downstream scRNA-seq analysis. We apply validated, platform-appropriate pipelines with tumour-specific QC considerations — accounting for the high mitochondrial content of stressed tumour cells, the transcriptional complexity of immune infiltrates, and the batch effects inherent in multi-patient oncology datasets.
- Raw data processing and alignment — Cell Ranger, STARsolo, and Alevin-fry processing for 10x Genomics, BD Rhapsody, and Parse Biosciences platforms; UMI counting, barcode filtering, and intronic read handling for nascent RNA capture
- Tumour-specific QC — Adaptive per-sample QC thresholds accounting for tumour cell heterogeneity; mitochondrial gene fraction assessment with context-aware filtering; SoupX and CellBender ambient RNA removal; Scrublet and DoubletFinder doublet detection
- Batch correction and multi-sample integration — Harmony, scVI, and BBKNN-based batch correction across patients, timepoints, and tissue sites; reference-based integration with healthy tissue atlases; patient-level pseudobulk aggregation for differential expression
- Cell type annotation — Automated annotation with SingleR, CellTypist, and scType against cancer-specific reference atlases; manual marker-based annotation of major immune, stromal, and malignant cell populations; cell type confidence scoring and ambiguous cell filtering
2. Tumour Microenvironment (TME) Immune Profiling T Cell · Macrophage · NK · DC · Treg · Exhaustion
The immune composition and functional state of the tumour microenvironment is a primary determinant of immunotherapy response, tumour immune evasion, and patient prognosis. We provide comprehensive, high-resolution TME immune profiling — characterising every major immune population and their functional states from scRNA-seq data with direct translational relevance.
- T cell subset and exhaustion profiling — High-resolution CD8+ and CD4+ T cell subtype annotation (naive, stem cell memory, central memory, effector memory, tissue-resident, terminally exhausted); exhaustion scoring with TOX, PDCD1, LAG3, HAVCR2, TIGIT gene signatures; progenitor exhausted vs. terminally exhausted T cell distinction; CD4+ Treg identification and FOXP3/CTLA4 expression profiling
- Tumour-associated macrophage (TAM) polarisation — M1/M2-like TAM polarisation scoring; SPP1+ and C1Q+ TAM subtype identification; monocyte-to-macrophage differentiation trajectory analysis; cross-tumour TAM state comparison against published pan-cancer TAM atlases
- NK cell, dendritic cell, and B cell profiling — NK cell cytotoxicity and exhaustion scoring; conventional DC1 and DC2, plasmacytoid DC, and mature DC subtype identification; tumour-infiltrating B cell and plasma cell characterisation; tertiary lymphoid structure (TLS) gene signature scoring
- TME composition and immune desert/excluded/inflamed classification — Immune cell fraction quantification per tumour sample; immune phenotype classification (desert, excluded, inflamed) from spatial and scRNA-seq evidence; correlation of TME composition with clinical response and survival outcomes
3. Cancer Cell State & Clonal Evolution Analysis InferCNV · CopyKAT · Stemness · EMT · Plasticity
Malignant cells within a single tumour exist in a spectrum of transcriptional states — ranging from cycling progenitor-like cells to differentiated, senescent, or epithelial-mesenchymal transition (EMT) states — and evolve under therapeutic selection through both genetic and non-genetic mechanisms. We provide expert analysis of cancer cell heterogeneity, transcriptional plasticity, and genomic clonal evolution from scRNA-seq data.
- Malignant cell identification and copy number inference — InferCNV, CopyKAT, and SCEVAN-based copy number alteration inference from scRNA-seq to distinguish malignant from normal cells; tumour ploidy estimation; chromosomal gain and loss mapping at single-cell resolution
- Cancer cell transcriptional state mapping — Non-negative matrix factorisation (NMF) and gene programme identification across cancer cells; mapping of cancer cell states against published pan-cancer meta-programmes (proliferating, stress, EMT, differentiated, hypoxic); cancer stem cell and tumour-initiating cell gene signature scoring
- Epithelial-mesenchymal transition (EMT) analysis — Continuous EMT scoring along epithelial-hybrid-mesenchymal axes; partial EMT state identification; EMT programme association with invasion, metastasis, and therapy resistance phenotypes
- Clonal evolution and therapy resistance tracking — Longitudinal scRNA-seq comparison of pre- and post-treatment tumours; identification of therapy-resistant cell states pre-existing before treatment vs. induced by treatment; clonal selection and transcriptional reprogramming under drug pressure; PyClone-VI integration for genetic clonal dynamics
4. Cell-Cell Communication & Trajectory Analysis CellChat · NicheNet · Monocle3 · RNA Velocity
Tumour progression, immune evasion, and therapy resistance emerge from dynamic intercellular signalling networks within the TME — and from the differentiation trajectories of both malignant and immune cell populations over time. We provide expert ligand-receptor interaction analysis and pseudotime trajectory modelling to reveal the signalling logic of tumour biology.
- Ligand-receptor and cell-cell communication analysis — CellChat, NicheNet, and LIANA-based intercellular signalling network inference; identification of tumour cell-to-immune cell immunosuppressive signals (TGF-β, VEGF, PD-L1/PD-1, TIGIT/CD155); cancer-associated fibroblast (CAF) signalling to immune and tumour cells; altered communication networks between responders and non-responders
- Pseudotime and differentiation trajectory analysis — Monocle3 and PAGA-based differentiation trajectory reconstruction; T cell exhaustion progression trajectory; cancer cell EMT and stemness acquisition trajectories; identification of branching decision points in tumour cell fate
- RNA velocity and transcriptional dynamics — scVelo and Dynamo-based RNA velocity analysis; spliced/unspliced ratio modelling for cell state transition directionality; identification of actively transitioning cell populations undergoing fate changes in the TME
- Gene regulatory network inference — SCENIC and pySCENIC transcription factor regulon activity scoring; identification of master regulators driving immune exhaustion, cancer stemness, or EMT transcriptional programmes; drug target identification from regulon disruption analysis
5. Multi-Omics Integration & Spatial Transcriptomics CITE-seq · Multiome · Visium · Xenium · Spatial TME
The full biological complexity of tumour microenvironments is best captured by integrating single-cell transcriptomics with protein expression, chromatin accessibility, and spatial localisation data — revealing not just what cells are present, but where they are, what they look like at the protein level, and how their chromatin landscape shapes their transcriptional identity.
- CITE-seq protein and transcriptome co-profiling — DSB and CLR-based ADT normalisation; joint protein-RNA clustering for high-confidence immune cell phenotyping; surface receptor expression validation against transcriptomic data; flow cytometry-equivalent immunophenotyping at single-cell resolution
- 10x Multiome ATAC + Gene Expression — Joint chromatin accessibility and transcriptome analysis; peak-to-gene linkage and transcription factor footprinting in cancer and immune cells; identification of epigenetically silenced tumour suppressor genes and open chromatin in therapy-resistant states
- Spatial transcriptomics of tumour tissue — Visium and Visium HD analysis of FFPE and fresh-frozen tumour sections; cell type deconvolution with cell2location and RCTD; spatial mapping of immune exclusion zones, tumour invasion fronts, and TLS structures; Xenium and MERSCOPE single-cell resolution spatial TME profiling
- Integration with bulk omics and clinical data — Bulk RNA-seq deconvolution with CIBERSORTx and MuSiC using scRNA-seq-derived signatures; integration of scRNA-seq cell state scores with WGS somatic mutation data; correlation of single-cell TME features with clinical response, progression-free survival, and overall survival
Key Applications
scRNA-seq oncology bioinformatics across tumour types, therapeutic modalities, and translational research contexts.
- Immune checkpoint inhibitor response and resistance TME profiling
- CAR-T and TIL therapy target antigen and TME barrier identification
- Pre- and post-treatment biopsy comparison for therapy resistance mechanisms
- Cancer stem cell and tumour-initiating cell transcriptional characterisation
- Pan-cancer TME atlas construction and cross-tumour immune comparison
- Tumour metastasis and EMT transcriptional programme analysis
- Single-cell biomarker discovery for immunotherapy patient stratification
- Spatial mapping of immune exclusion, TLS, and tumour invasion architecture
Tools, Technologies & Reference Resources
Validated, cutting-edge single-cell bioinformatics tools and all major oncology single-cell reference resources.
- Processing: Cell Ranger, STARsolo, Alevin-fry, SoupX, CellBender, Scrublet
- Analysis: Seurat, Scanpy, scVI, Harmony, BBKNN, SingleR, CellTypist
- CNV Inference: InferCNV, CopyKAT, SCEVAN, numbat
- Trajectory: Monocle3, scVelo, PAGA, Dynamo, CellRank, Palantir
- Cell Communication: CellChat, NicheNet, LIANA, CellPhoneDB
- Regulons: SCENIC, pySCENIC, decoupleR, DoRothEA
- Spatial: SpaceRanger, Squidpy, cell2location, RCTD, Giotto, BayesSpace
- Multi-omics: ArchR, Signac, WNN (Seurat), totalVI, MultiVI
- Bulk Deconvolution: CIBERSORTx, MuSiC, EPIC, BayesPrism
- Human Cell Atlas / TISCH2 — Single-cell tumour immune reference atlases for TME annotation
- pan-cancer scRNA-seq atlases — TCGA, GEO, and published pan-cancer single-cell datasets for cross-study integration
- MSigDB / CancerSEA — Cancer gene signatures for cell state scoring and pathway analysis
Project Deliverables
Structured, publication-ready scRNA-seq oncology bioinformatics outputs for every project.
- Annotated single-cell object (Seurat/AnnData) with cell type labels, QC metrics, and embeddings
- UMAP visualisations with cell type, gene expression, and clinical metadata overlays
- TME composition tables per sample with immune cell subset fractions and statistical comparisons
- Differential expression and gene set enrichment results between conditions of interest
- Cancer cell CNV inference plots and malignant vs. normal cell classification
- Trajectory and pseudotime analysis outputs with lineage plots and driver gene identification
- Cell-cell communication network figures and ligand-receptor interaction tables
- Publication-ready figures (PDF/SVG/PNG at 300 dpi)
- Full written scientific report with methods, results, biological interpretation, and translational context
- Spatial transcriptomics TME mapping and cell type deconvolution
- CITE-seq protein co-expression and multimodal clustering analysis
- Integration with patient WGS somatic mutation and clinical outcome data
- Bulk RNA-seq deconvolution using scRNA-seq-derived cell type signatures
- Manuscript methods section and supplementary figure legends
- Grant application single-cell oncology sections and preliminary data
- Long-term retainer for ongoing cohort expansion and dataset integration
Frequently Asked Questions
Common questions from cancer research groups, translational oncology teams, and pharmaceutical drug discovery programmes.
For robust TME immune profiling, we generally recommend capturing at least 5,000–10,000 cells per sample, with a minimum of 5–6 biological replicates (patient samples) per condition to enable statistically meaningful comparisons. Rarer immune populations such as plasmacytoid DCs, NK cells, and plasma cells may require deeper sequencing or larger cell numbers to capture reliably. We advise on study design, expected cell yield, and sequencing depth at project scoping — before samples are collected or sequenced.
Yes. We use copy number alteration inference tools — InferCNV, CopyKAT, and SCEVAN — to infer chromosomal gains and losses from scRNA-seq expression data, which distinguishes aneuploid malignant cells from diploid stromal and immune cells. This approach works without matched normal tissue, although having a reference normal cell population (immune or stromal cells within the same tumour sample) significantly improves inference accuracy. For haematological malignancies, we use alternative approaches based on known malignant cell markers and clonotypic BCR/TCR sequences.
Yes — with important caveats. Fresh or frozen tissue is strongly preferred for scRNA-seq, as FFPE fixation degrades RNA and dramatically reduces cell viability and gene detection rates. However, Visium HD and 10x Xenium spatial transcriptomics platforms are specifically optimised for FFPE tissue — enabling spatially-resolved gene expression profiling from archival FFPE tumour blocks. We advise on the most appropriate platform and analytical approach for your specific sample type and research question.
Yes. Single-cell-derived cell type signatures can be used to estimate immune and stromal cell type fractions from bulk RNA-seq data using deconvolution tools such as CIBERSORTx, MuSiC, BayesPrism, and EPIC. This is particularly powerful for leveraging large, clinically annotated bulk RNA-seq cohorts (TCGA, GEO, clinical trial datasets) to validate TME findings from scRNA-seq in statistically powered patient populations with survival outcome data.
Absolutely. We assist with the single-cell bioinformatics and computational oncology sections of grant applications — including proposed scRNA-seq analysis workflows, TME profiling methodology, clonal evolution approaches, and preliminary single-cell data. Please contact us as early as possible in the grant preparation process to allow time for any preliminary analyses that would strengthen the application.
Related Research Areas & Services
scRNA-seq oncology bioinformatics connects to multiple complementary services we support.
- Cancer & Oncogenomics — Somatic variant calling, mutational signature analysis, TMB and MSI scoring, neoantigen prediction, and WGS-based clonal evolution for integration with scRNA-seq tumour profiling
- Immunology & Immuno-Oncology — Immune cell biology, checkpoint pathway analysis, neoantigen identification, and TCR/BCR repertoire analysis to complement single-cell TME profiling
- Immune Repertoire (TCR/BCR) — Paired scTCR-seq and scBCR-seq clonotype-phenotype integration with scRNA-seq TME data for tumour-reactive clone identification
- Cell & Gene Therapy Bioinformatics — CAR-T and TIL product transcriptomic characterisation, exhaustion profiling, and TME interaction analysis for adoptive cell therapy programmes
- Drug Development & AI-Driven Discovery — Single-cell-informed biomarker discovery, patient stratification, and TME-based drug target identification for immuno-oncology drug development
- Custom Software & Pipeline Development — Bespoke scRNA-seq analysis platforms, interactive single-cell atlas explorers, and automated tumour profiling pipelines for internal research teams
Ready to Unlock the Full Biology of Your Tumour Data?
Tell us about your tumour type, your scRNA-seq data, and your research or translational objectives. Our single-cell oncology bioinformatics team will design a tailored analytical plan — typically within 48 hours of your enquiry. Whether you need TME immune profiling, cancer cell state mapping, clonal evolution analysis, therapy resistance characterisation, or spatial transcriptomics of tumour sections, we are here to deliver expert, publication-ready single-cell results from day one.
