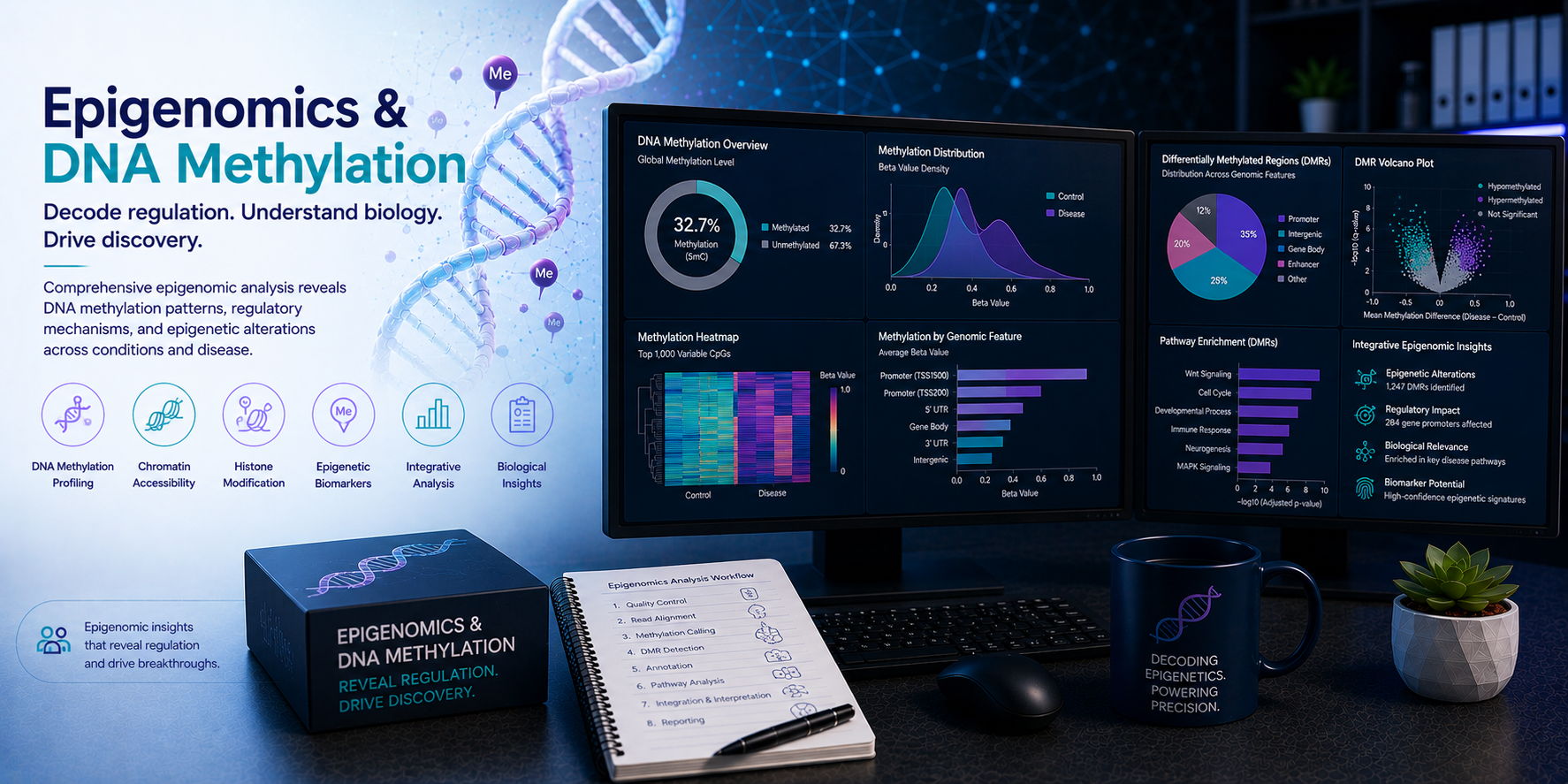
Epigenomics — the genome-wide study of DNA methylation, chromatin accessibility, histone modifications, and three-dimensional genome organisation — provides a critical layer of biological information between genotype and phenotype. Epigenetic marks are dynamic, tissue-specific, and disease-associated; they regulate gene expression without altering the underlying DNA sequence; and they encode the developmental history, environmental exposures, and disease state of a cell in ways that the genome alone cannot reveal. From Illumina methylation arrays and whole-genome bisulphite sequencing to ATAC-seq chromatin accessibility profiling and ChIP-seq histone modification mapping, epigenomics generates rich, complex datasets demanding specialist bioinformatics analysis. At BioinformaticsNext, we provide expert epigenomics and DNA methylation bioinformatics — supporting cancer research, developmental biology, clinical epigenetics, drug discovery, and cell and gene therapy programmes with rigorous, reproducible, and biologically interpretable epigenomic analysis.
Epigenomics & DNA Methylation Bioinformatics: Methylation Arrays, Chromatin Accessibility & Histone Modifications
Expert bioinformatics for Illumina EPIC methylation arrays, WGBS, ATAC-seq, ChIP-seq, CUT&RUN, and multi-modal epigenomics integration — across cancer epigenetics, developmental biology, clinical epigenomics, and regulatory genomics applications.
The epigenome is not static — it changes dynamically during development, differentiation, ageing, and disease. Cancer epigenomes show global hypomethylation alongside focal promoter hypermethylation that silences tumour suppressor genes; stem cells maintain bivalent chromatin domains poised for activation or repression; immune cells undergo rapid chromatin remodelling during activation; and disease-associated epigenetic changes accumulate in response to environmental exposures and ageing. Understanding these epigenomic dynamics requires not just measuring methylation or chromatin state at a single timepoint but integrating epigenomic data with transcriptomics, genomics, and clinical metadata to extract mechanistic and translational insight. At BioinformaticsNext, we provide the full epigenomics bioinformatics stack — from array and sequencing data processing through differential methylation, chromatin accessibility, regulatory element identification, and multi-omics epigenomic integration.
What We Support
Comprehensive epigenomics and DNA methylation bioinformatics across all major platforms, assay types, and biological applications.
- Illumina 450K, EPIC (850K), and EPICv2 methylation array processing and differential analysis
- Whole-genome bisulphite sequencing (WGBS) and reduced representation bisulphite sequencing (RRBS)
- ATAC-seq and FAIRE-seq chromatin accessibility profiling and differential peak analysis
- ChIP-seq for histone modifications (H3K27ac, H3K4me3, H3K27me3, H3K9me3) and TF binding
- CUT&RUN and CUT&TAG high-resolution chromatin profiling
- Enhancer and regulatory element identification and activity mapping
- Epigenetic clock and biological age estimation from methylation data
- DNA methylation-based tumour classification and tissue-of-origin analysis
- Multi-omics epigenomics integration with transcriptomics and genomics
- Single-cell ATAC-seq and scMultiome epigenomics analysis
Our Epigenomics & DNA Methylation Bioinformatics Services
Specialist epigenomics bioinformatics — from methylation array and bisulphite sequencing processing through chromatin accessibility, histone modification, regulatory element analysis, and clinical epigenomics applications.
All analyses are tailored to your epigenomics platform, tissue type, experimental design, and biological, clinical, or drug discovery objectives.
1. DNA Methylation Array Analysis EPIC · 450K · EPICv2 · minfi · ChAMP · Differential Methylation
Illumina methylation arrays — including the 450K, EPIC (850K), and EPICv2 arrays — provide cost-effective, high-throughput genome-wide DNA methylation profiling at CpG resolution, and remain the most widely used platform for clinical epigenomics, epigenetic clock analysis, cell-type deconvolution, and cancer epigenomics research. We apply validated, clinically tested array processing pipelines with appropriate QC, normalisation, and statistical analysis.
- Array QC and normalisation — minfi and ChAMP-based Illumina EPIC and 450K array processing; sample quality assessment from bisulphite conversion efficiency, detection p-value filtering, and sex concordance; BMIQ, NOOB, and functional normalisation strategies; cross-batch normalisation and ComBat correction for multi-cohort analysis; cross-array platform harmonisation (450K to EPIC) with manifest liftover
- Differential methylation analysis — limma and DSS-based differentially methylated position (DMP) identification; dmrseq and bumphunter differentially methylated region (DMR) calling; cell-type composition correction with Houseman/CETYGO for blood and tissue arrays; covariate adjustment for age, sex, batch, and smoking; effect size (delta beta) and clinical significance thresholding
- Epigenetic clock and biological age estimation — Horvath, Hannum, PhenoAge, GrimAge, DunedinPACE, and PCClocks biological age calculation; epigenetic age acceleration analysis and association with clinical phenotypes; multi-clock concordance assessment; epigenetic age as a pharmacological response biomarker
- Cell-type deconvolution and composition analysis — Houseman, CETYGO, and EpiDISH reference-based cell-type fraction estimation from blood, PBMC, and tissue arrays; immune cell composition comparison between disease groups; cell-type deconvolution validation against matched flow cytometry data
2. Whole-Genome Bisulphite Sequencing & RRBS Analysis Bismark · WGBS · RRBS · CpG Context · DMR Calling
Whole-genome bisulphite sequencing (WGBS) and reduced representation bisulphite sequencing (RRBS) provide base-resolution DNA methylation quantification across CpG, CHG, and CHH contexts — enabling comprehensive methylome characterisation beyond the CpG sites covered by arrays, with applications in plant epigenomics, imprinting analysis, and high-resolution regulatory element methylation mapping.
- Bisulphite sequencing data processing — Bismark and bsseq-based bisulphite read alignment to reference genome; non-conversion rate estimation and sample QC; per-CpG methylation extraction in CpG, CHG, and CHH contexts; coverage filtering and smoothing; methylation matrix construction across samples and conditions
- Differential methylation analysis from WGBS — DSS and MethylKit DMP and DMR calling from WGBS data; Wald test and beta-binomial model-based differential testing; DMR size, mean methylation difference, and coverage-weighted significance reporting; annotation of DMRs to CpG islands, shores, shelves, and gene regulatory elements
- CpG island and regulatory element methylation — CpG island, shore, and shelf methylation profiling; promoter, enhancer, gene body, and intergenic region methylation context analysis; bivalent domain identification from combined H3K4me3 and H3K27me3 data; transcription factor binding site methylation interference analysis
- Imprinting and allele-specific methylation — Allele-specific methylation (ASM) detection from WGBS data using SNP-methylation phasing; imprinted gene methylation analysis; parent-of-origin ASM identification from trio data; epigenetic mutation and stochastic epigenetic variation analysis
3. ATAC-seq & Chromatin Accessibility Analysis ATAC-seq · MACS2 · ChromVAR · Peak Annotation · TF Footprinting
ATAC-seq (Assay for Transposase-Accessible Chromatin with sequencing) profiles genome-wide open chromatin — identifying active regulatory elements including promoters, enhancers, insulators, and transcription factor binding sites from as few as 500–50,000 cells. We provide comprehensive ATAC-seq bioinformatics from read alignment and peak calling through differential accessibility, TF footprinting, and integration with gene expression data.
- ATAC-seq data processing and QC — Trimmomatic and Trim Galore adapter trimming; Bowtie2 alignment to reference genome with mitochondrial and blacklist region removal; ATAC-seq-specific QC: TSS enrichment score, nucleosomal banding pattern (sub-nucleosomal, mono-, di-, tri-nucleosomal), fraction of reads in peaks (FRiP), and library complexity estimation
- Peak calling and annotation — MACS2 and Genrich narrow peak calling from ATAC-seq data; consensus peak atlas construction from multi-sample peak sets with DiffBind; peak annotation to genomic features (promoter, intragenic, intergenic, enhancer) with ChIPseeker and HOMER; peak distance to TSS and nearest gene assignment
- Differential chromatin accessibility analysis — DESeq2 and edgeR differential peak accessibility between conditions, timepoints, and cell types; DiffBind consensus peak differential analysis; volcano plot and MA plot visualisation; GO and pathway enrichment of genes near differentially accessible peaks
- Transcription factor activity and footprinting — ChromVAR TF motif deviation scoring for genome-wide TF activity inference; TOBIAS and HINT-ATAC TF footprinting from ATAC-seq for active TF binding site identification; JASPAR and ENCODE motif database enrichment analysis in differential peaks; TF binding site conservation across cell types and conditions
4. ChIP-seq, CUT&RUN & Histone Modification Profiling H3K27ac · H3K4me3 · Enhancers · Super-Enhancers · ENCODE
ChIP-seq and its successors CUT&RUN and CUT&TAG provide genome-wide maps of histone modifications and transcription factor occupancy — defining the active and repressed regulatory landscape of a cell. Active promoters (H3K4me3, H3K27ac), poised and active enhancers (H3K4me1, H3K27ac), bivalent domains (H3K4me3 + H3K27me3), constitutive heterochromatin (H3K9me3), and Polycomb-repressed chromatin (H3K27me3) each carry distinct biological interpretations that we characterise with validated bioinformatics pipelines.
- ChIP-seq and CUT&RUN data processing — Trim Galore and Bowtie2 alignment; ENCODE blacklist filtering; Picard duplicate removal; spike-in normalisation (ERCC, Drosophila) for quantitative ChIP-seq comparison; deepTools bamCoverage bigWig track generation; ChIP quality metrics: SSD, NSC, RSC, and IDR peak reproducibility assessment
- Peak calling and histone mark annotation — MACS2 narrow peak calling for sharp marks (H3K4me3, TF binding) and broad peak calling for diffuse marks (H3K27me3, H3K9me3); SEACR peak calling for CUT&RUN data; ChromHMM and Segway chromatin state segmentation from multi-mark ChIP-seq; ENCODE reference chromatin state annotation comparison
- Enhancer and super-enhancer identification — H3K27ac and H3K4me1-based active enhancer identification; ROSE super-enhancer calling from H3K27ac signal; tissue-specific enhancer activity comparison; enhancer-gene linkage from 3D chromatin interaction data or distance-based assignment; differential enhancer activity between conditions
- Differential histone modification analysis — DESeq2-based differential histone modification between conditions; DiffBind consensus peak differential analysis; bivalent domain resolution during differentiation; PRC2 target identification from H3K27me3 coverage; histone modification co-occurrence and co-exclusion analysis
5. Clinical Epigenomics, Tumour Classification & Multi-Omics Integration Tumour Classifier · Epigenetic Biomarkers · scATAC-seq · MOFA+
Clinical epigenomics harnesses DNA methylation as a diagnostic and prognostic tool — with brain tumour methylation classifiers now used in routine neuropathology, cfDNA methylation enabling multi-cancer early detection, and epigenetic biomarkers predicting immunotherapy response. We provide specialist bioinformatics for clinical epigenomics applications and multi-omics epigenomic integration.
- DNA methylation-based tumour classification — Illumina EPIC array-based brain tumour methylation classification using Capper et al. random forest classifier; UMAP-based methylation profile clustering against reference cohorts; copy number profiling from methylation array intensity data using conumee; tumour purity and cell-type composition assessment from methylation data
- cfDNA methylation and epigenetic liquid biopsy — Plasma cfDNA methylation profiling for tissue-of-origin deconvolution using CelFiE and MethAtlas; cancer-associated differentially methylated region (DMR) signature scoring; methylation-based multi-cancer early detection model development; cfDNA methylation biomarker validation across independent clinical cohorts
- Single-cell ATAC-seq and scMultiome analysis — ArchR and Signac-based scATAC-seq processing, LSI dimensionality reduction, and cell type clustering; 10x Multiome joint ATAC+RNA analysis; gene activity score calculation; peak-to-gene linkage from co-accessibility and co-expression; TF motif enrichment per cell type and disease state
- Multi-omics epigenomics integration — MOFA+ latent factor integration of methylation, ATAC-seq, RNA-seq, and ChIP-seq data; epigenomics-transcriptomics correlation at the gene and regulatory element level; methylation QTL (mQTL) and chromatin accessibility QTL (caQTL) mapping for GWAS variant functional annotation; regulatory element activity-expression linkage analysis
Key Applications
Epigenomics and DNA methylation bioinformatics across cancer, developmental biology, clinical diagnostics, and drug discovery.
- Cancer promoter hypermethylation and tumour suppressor silencing analysis
- Brain tumour methylation-based classification for neuropathology
- Epigenetic clock biological age estimation in ageing and disease cohorts
- Enhancer and super-enhancer mapping in cancer and normal cells
- ATAC-seq chromatin remodelling in drug response and resistance
- cfDNA methylation multi-cancer early detection biomarker development
- Single-cell epigenomics for cell type-resolved regulatory element mapping
- Epigenetic drug target identification from chromatin accessibility profiling
Tools, Technologies & Reference Databases
Validated, widely adopted epigenomics bioinformatics tools and all major epigenomic reference resources.
- Array Processing: minfi, ChAMP, SeSAMe, ENmix, methylumi, wateRmelon
- WGBS/RRBS: Bismark, bsseq, DSS, MethylKit, MethylDackel, Trim Galore
- ATAC-seq: Bowtie2, MACS2, Genrich, DiffBind, ChromVAR, TOBIAS, ChIPseeker
- ChIP-seq/CUT&RUN: deepTools, SEACR, ROSE, ChromHMM, IDR, HOMER, ENCODE pipeline
- Single-Cell Epigenomics: ArchR, Signac, SnapATAC2, EpiScanpy, scATAC-pro
- ENCODE / Roadmap Epigenomics — Reference histone modification and chromatin accessibility maps across human cell types and tissues for comparative epigenomics
- UCSC / Ensembl Regulatory Build — Annotated regulatory elements, CpG islands, and genome segmentation references for peak annotation
- JASPAR / ENCODE TF Motifs — Transcription factor binding motif databases for ChromVAR and footprinting analysis
- GEO / NCBI SRA / BLUEPRINT — Public epigenomics reference datasets for comparative analysis and cell-type deconvolution reference construction
- MethAtlas / CelFiE / Houseman Reference — Cell-type and tissue methylation reference atlases for deconvolution and tissue-of-origin analysis
Project Deliverables
Structured, publication-ready epigenomics and DNA methylation bioinformatics outputs for every project.
- Array or sequencing QC report with bisulphite conversion efficiency, coverage, and sample pass/fail
- Normalised methylation beta/M-value matrix or per-CpG methylation coverage files
- DMP and DMR tables with delta beta, adjusted p-values, and genomic feature annotation
- ATAC-seq or ChIP-seq peak atlas with annotation to regulatory elements and nearest genes
- Differential accessibility or histone modification results with effect sizes and FDR
- TF motif enrichment and ChromVAR activity deviation scores per condition
- Epigenetic clock biological age estimates and acceleration statistics (array projects)
- Publication-ready figures (PDF/SVG/PNG at 300 dpi): heatmaps, genome browser tracks, volcano plots, UMAPs
- Full written scientific report with methods, results, biological interpretation, and recommendations
- Brain tumour methylation classification report with UMAP cluster visualisation
- cfDNA methylation tissue-of-origin deconvolution and cancer detection scoring
- Single-cell ATAC-seq cell type annotation and regulatory element mapping
- Multi-omics epigenomics integration with RNA-seq and genomics data
- Super-enhancer calling and enhancer-gene linkage analysis
- Manuscript methods section and supplementary figure legends
- Grant application epigenomics sections and preliminary data
- Long-term retainer for ongoing epigenomics programme analytical support
Frequently Asked Questions
Common questions from cancer epigenetics researchers, clinical genomics groups, and drug discovery teams.
Illumina EPIC (850K) and EPICv2 arrays measure methylation at approximately 850,000 and 935,000 pre-selected CpG sites — providing excellent coverage of gene promoters, CpG islands, and regulatory regions at relatively low cost, making them ideal for large clinical cohort studies, epigenetic clocks, and cancer epigenomics. WGBS sequences every CpG in the genome (approximately 28 million in humans) at base resolution — providing comprehensive coverage including non-CpG contexts (CHG, CHH relevant for plant and stem cell epigenomics), imprinted regions, repeat elements, and CpGs not covered by arrays, but at substantially higher cost and computational complexity. For clinical cohort studies and biomarker discovery, EPIC arrays are typically preferred; for mechanistic studies requiring complete methylome coverage, allele-specific methylation, or plant epigenomics, WGBS is essential.
DNA methylation in blood samples reflects the composite signal from multiple cell types — granulocytes, monocytes, B cells, T cells, NK cells, and erythrocyte precursors — and differences in cell-type composition between disease and control samples can confound differential methylation analysis if not corrected. We apply reference-based cell-type deconvolution using the Houseman, CETYGO, and EpiDISH algorithms with validated blood cell-type methylation reference profiles to estimate cell-type fractions per sample, and include these estimated fractions as covariates in the limma/DSS differential methylation model. We validate deconvolution estimates against matched complete blood count data where available.
Chromatin accessibility refers to the physical openness of chromatin — regions of the genome where nucleosomes are displaced or repositioned to allow transcription factor binding and gene regulatory activity. Open chromatin corresponds to active promoters, enhancers, insulators, and other cis-regulatory elements. ATAC-seq uses a hyperactive Tn5 transposase to preferentially insert sequencing adapters into accessible chromatin regions, generating a genome-wide map of open chromatin from as few as 500 cells. Because accessible chromatin changes rapidly in response to differentiation signals, drug treatment, and disease state, ATAC-seq is particularly powerful for studying regulatory chromatin dynamics — capturing changes in TF occupancy and enhancer activity that precede and drive transcriptional changes.
Yes. We provide comprehensive single-cell ATAC-seq bioinformatics using ArchR and Signac — including fragment file processing, TSS enrichment and library complexity QC, iterative LSI dimensionality reduction, graph-based clustering, cell type annotation from gene activity scores and marker peak enrichment, differential accessibility analysis between cell populations, ChromVAR TF motif deviation scoring, and peak-to-gene linkage analysis. For 10x Multiome ATAC+Gene Expression data, we perform joint RNA-ATAC integration with weighted nearest neighbour (WNN) clustering and transcription factor regulon inference with SCENIC+.
Absolutely. We assist with the epigenomics bioinformatics sections of grant applications — including proposed methylation array or WGBS analysis workflows, ATAC-seq chromatin accessibility methodology, ChIP-seq histone modification approaches, clinical epigenomics applications, and preliminary epigenomic data. Please contact us as early as possible in the grant preparation process to allow time for any preliminary analyses that would strengthen the scientific case.
Related Research Areas & Services
Epigenomics and DNA methylation bioinformatics connects to multiple complementary services we support.
- Cancer & Oncogenomics — Somatic variant calling, mutational signature analysis, and tumour genomics integration with cancer epigenomics for comprehensive tumour characterisation and epigenetic driver identification
- Single-Cell Multi-Omics (CITE-seq) — 10x Multiome ATAC+Gene Expression joint analysis, WNN integration, and SCENIC+ gene regulatory network inference from single-cell epigenomics data
- Drug Development & AI-Driven Discovery — Epigenetic drug target identification from chromatin profiling, ATAC-seq response to epigenetic inhibitor treatment, and methylation biomarker development for clinical programmes
- Multi-Omics Integration — MOFA+ epigenomics-transcriptomics-genomics integration, mQTL and caQTL mapping for GWAS variant functional annotation, and regulatory element activity-expression linkage analysis
- Long-Read Sequencing (ONT/PacBio) — Direct 5mC and 5hmC methylation detection from Oxford Nanopore sequencing without bisulphite conversion, and allele-specific methylation from phased long-read data
- Custom Software & Pipeline Development — Bespoke epigenomics analysis platforms, automated methylation reporting pipelines, and interactive genome browser track generation for internal research and clinical teams
Ready to Advance Your Epigenomics Research?
Tell us about your epigenomics platform, your tissue or cell type, your experimental design, and your biological, clinical, or drug discovery objectives. Our epigenomics and DNA methylation bioinformatics team will design a tailored analytical plan — typically within 48 hours of your enquiry. Whether you need methylation array differential analysis and epigenetic clock estimation, WGBS base-resolution methylome profiling, ATAC-seq chromatin accessibility and TF footprinting, ChIP-seq histone modification mapping, brain tumour methylation classification, or single-cell epigenomics, we are here to deliver expert, publication-ready epigenomics results from day one.
