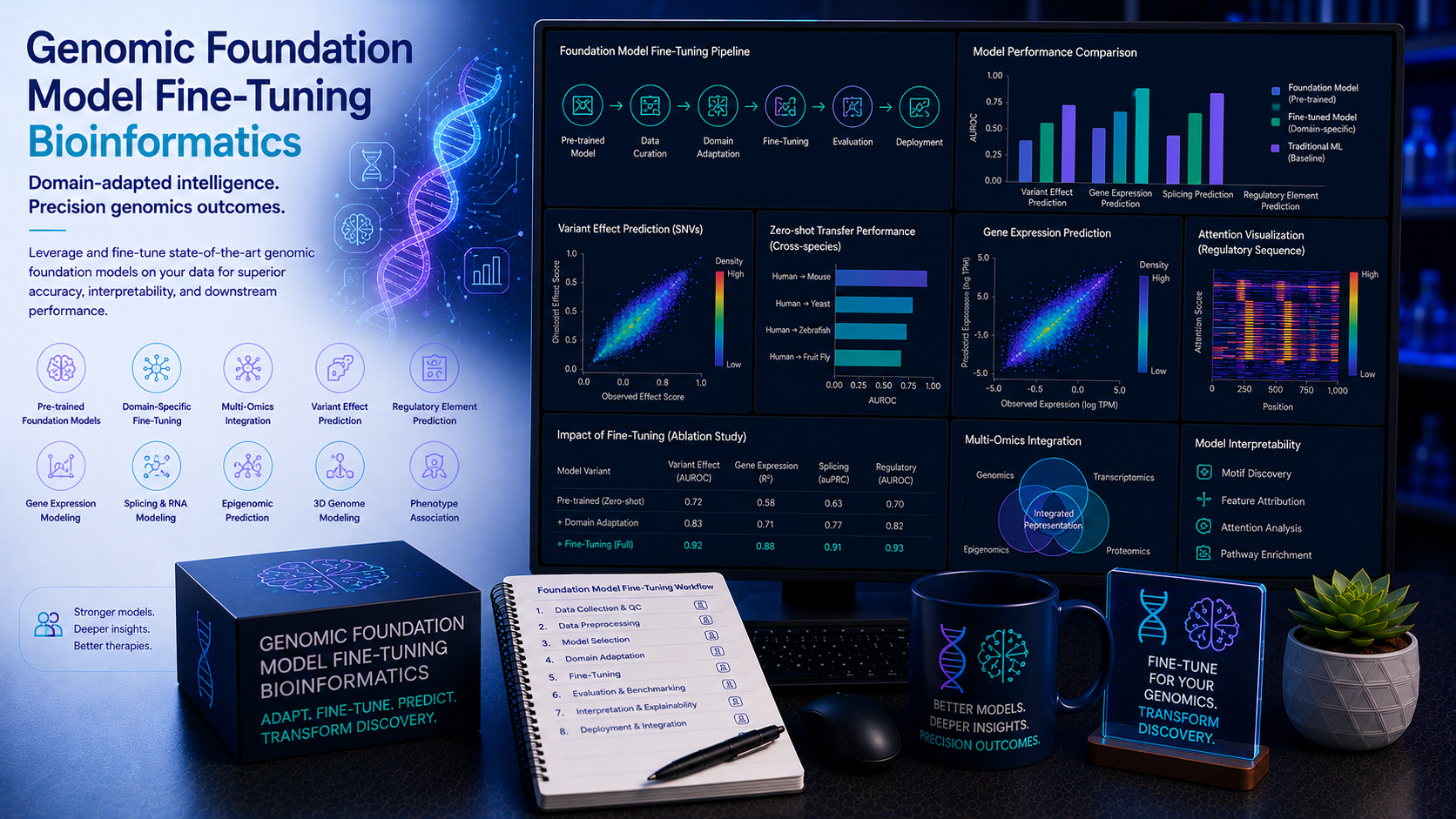
Genomic foundation models — large-scale transformer and attention-based neural networks pre-trained on vast quantities of DNA, RNA, and protein sequences — are redefining the boundaries of computational biology. Models such as AlphaGenome, DNABERT-2, Nucleotide Transformer, HyenaDNA, Enformer, Evo, and ESM-3 have learned rich, generalisable representations of genomic sequence that can be fine-tuned for specific downstream tasks with a fraction of the labelled data required to train a task-specific model from scratch. From predicting the regulatory activity of non-coding variants and splice site disruption to functional annotation of novel genomic sequences, protein engineering, and RNA structure prediction, genomic foundation models are accelerating biological discovery at every scale. At BioinformaticsNext, we provide specialist genomic foundation model fine-tuning and deployment services — supporting academic research groups, pharmaceutical companies, and genomics technology companies in applying state-of-the-art genomic LLMs to their specific biological questions with rigorous validation and production-ready deployment.
Genomic Foundation Model Fine-Tuning: AlphaGenome, DNABERT, Nucleotide Transformer & Genomic LLMs
Expert fine-tuning, evaluation, and deployment of genomic foundation models — including AlphaGenome, DNABERT-2, Nucleotide Transformer, HyenaDNA, Enformer, Evo, and ESM-3 — for variant effect prediction, regulatory genomics, protein function, RNA biology, and clinical genomics applications.
The genomic foundation model landscape has evolved rapidly — from DNABERT's pioneering application of BERT-style masked language modelling to DNA sequences through Enformer's long-range regulatory sequence prediction, HyenaDNA's ultra-long context DNA modelling, the Nucleotide Transformer's multi-species pre-training, and the recent release of AlphaGenome for genome-wide regulatory prediction and Evo for cross-modal DNA-RNA-protein modelling. Each model has distinct architectural strengths, pre-training data characteristics, and optimal application domains — and selecting, fine-tuning, and validating the right model for a specific genomic prediction task requires deep expertise in both the biology and the machine learning.
At BioinformaticsNext, we provide end-to-end genomic foundation model services — from model selection and dataset curation through supervised fine-tuning, parameter-efficient adaptation (LoRA, adapter layers), rigorous benchmarking against task-specific baselines, and production deployment in cloud or on-premise environments — for any genomic sequence prediction task.
What We Support
Comprehensive genomic foundation model fine-tuning and deployment across DNA, RNA, and protein sequence modelling tasks and biological applications.
- AlphaGenome fine-tuning for genome-wide regulatory element and variant effect prediction
- DNABERT-2 and Nucleotide Transformer fine-tuning for genomic sequence classification tasks
- HyenaDNA and Caduceus fine-tuning for ultra-long context genomic sequence modelling
- Enformer adaptation for cis-regulatory sequence activity and gene expression prediction
- Evo fine-tuning for cross-modal DNA, RNA, and protein function prediction
- ESM-3 and ESM-2 protein language model fine-tuning for protein function and engineering
- Parameter-efficient fine-tuning (LoRA, QLoRA, adapters) for resource-constrained deployment
- Custom genomic training dataset curation, augmentation, and quality control
- Model benchmarking, interpretability analysis, and attention visualisation
- Production-ready model deployment on cloud infrastructure (AWS, GCP, Azure, HPC)
Our Genomic Foundation Model Services
End-to-end genomic foundation model fine-tuning — from model selection and dataset curation through supervised fine-tuning, parameter-efficient adaptation, rigorous benchmarking, and production deployment.
All projects are tailored to your genomic prediction task, training data availability, computational budget, deployment environment, and research or commercial objectives.
1. Model Selection & Task-Specific Architecture Design AlphaGenome · DNABERT-2 · HyenaDNA · Enformer · Evo · ESM-3
Selecting the right genomic foundation model for a specific prediction task is as important as fine-tuning it correctly. Different models have distinct architectural strengths, context window sizes, pre-training datasets, and performance profiles across task categories — and an ill-chosen foundation model will underperform regardless of fine-tuning quality. We provide expert model selection analysis and architecture design for every genomic LLM fine-tuning project.
- Foundation model landscape assessment — Systematic comparison of available genomic foundation models against your prediction task: AlphaGenome (genome-wide regulatory prediction, variant effect scoring), DNABERT-2 (general-purpose DNA sequence classification, multi-species), Nucleotide Transformer (6-mer tokenised transformer, benchmark tasks), HyenaDNA and Caduceus (ultra-long context up to 1Mb, rare variant modelling), Enformer (long-range gene expression prediction from regulatory sequences), Evo (DNA-RNA-protein cross-modal), ESM-3 and ESM-2 (protein sequences, protein engineering)
- Context window and tokenisation analysis — Sequence length and context window matching to prediction task requirements; k-mer vs. single-nucleotide tokenisation trade-offs; species-specific vs. multi-species pre-training data relevance assessment; model parameter scale vs. available fine-tuning data volume trade-off analysis
- Task head architecture design — Classification head design for sequence-to-label tasks (binary, multiclass, multilabel); regression head for continuous signal prediction; sequence-to-sequence architectures for variant effect scoring; token-level prediction heads for per-position annotation tasks; ensemble architectures combining multiple foundation models
- Baseline model comparison design — Pre-fine-tuning zero-shot and few-shot foundation model performance assessment; task-specific traditional ML baseline design (k-mer frequency, gapped k-mer SVM, DeepBind); benchmark dataset identification for rigorous performance comparison
2. Training Dataset Curation & Quality Control Dataset Curation · Class Balance · Augmentation · Leakage Prevention
The quality, representativeness, and proper partitioning of the fine-tuning dataset determines the ceiling of genomic foundation model performance far more than the choice of fine-tuning hyperparameters. We design and curate training datasets for genomic LLM fine-tuning with rigorous attention to sequence quality, class balance, biological relevance, and the prevention of data leakage between train, validation, and test splits.
- Genomic training data curation — Systematic curation of positive and negative training examples from ENCODE, Roadmap Epigenomics, JASPAR, ChIP-Atlas, EVE, ClinVar, gnomAD, and task-specific databases; reference genome version harmonisation (GRCh38, T2T-CHM13); sequence retrieval, flanking region specification, and duplicate removal
- Class balance and data augmentation — Positive-negative class ratio optimisation for imbalanced genomic datasets; reverse complement augmentation for strand-agnostic sequence tasks; species-aware data augmentation from homologous sequences; synthetic minority oversampling (SMOTE) adaptations for genomic feature spaces; uncertainty-weighted sampling for semi-supervised fine-tuning
- Train-validation-test splitting with leakage prevention — Chromosome-held-out splitting strategy to prevent sequence homology leakage between train and test sets; sequence identity clustering with MMseqs2 to prevent near-identical sequence data leakage in protein tasks; temporal holdout for variant datasets ensuring test variants postdate training data; benchmark alignment with established DNABERT and Nucleotide Transformer evaluation protocols
- Data quality and bias assessment — GC content distribution, repeat element contamination, and low-complexity sequence screening; cell-type and tissue representation bias assessment in epigenomic training data; population and ancestry representation assessment for variant-level training datasets; systematic bias identification and mitigation strategy design
3. Fine-Tuning, Parameter-Efficient Adaptation & Training LoRA · QLoRA · Adapters · Full Fine-Tuning · Multi-Task
Genomic foundation model fine-tuning ranges from lightweight parameter-efficient adaptation methods — that freeze most model weights and train only a small number of task-specific parameters — to full model fine-tuning where compute and data volume permit. We design and execute the optimal fine-tuning strategy for your prediction task, data availability, and computational budget.
- Full fine-tuning on GPU/HPC infrastructure — End-to-end supervised fine-tuning of DNABERT-2, Nucleotide Transformer, HyenaDNA, and Enformer on task-specific datasets using PyTorch and HuggingFace Transformers; learning rate scheduling (linear warmup with cosine decay, one-cycle); gradient accumulation for large effective batch sizes on limited GPU memory; mixed precision (FP16/BF16) and gradient checkpointing for memory efficiency
- Parameter-efficient fine-tuning (PEFT) — LoRA (Low-Rank Adaptation) and QLoRA (quantised LoRA) for resource-efficient adaptation of large genomic foundation models; adapter layer insertion at transformer blocks for multi-task fine-tuning with shared backbone; prefix tuning and prompt tuning for few-shot genomic sequence classification; rank and alpha hyperparameter optimisation for LoRA configurations
- Multi-task and transfer fine-tuning — Multi-task fine-tuning across related genomic prediction tasks sharing a backbone (e.g. simultaneous TF binding, chromatin accessibility, and histone modification prediction); sequential transfer fine-tuning from data-rich to data-sparse tasks; domain adaptive pre-training on task-relevant unlabelled genomic sequences before supervised fine-tuning
- Hyperparameter optimisation and training monitoring — Optuna and Weights & Biases-based learning rate, batch size, and dropout hyperparameter search; training loss and validation metric tracking; early stopping with model checkpoint selection; training stability assessment and gradient norm monitoring; compute-optimal training budget estimation
4. Benchmarking, Evaluation & Interpretability AUROC · Calibration · Attention · In-Silico Mutagenesis · SHAP
Rigorous, independent evaluation of fine-tuned genomic foundation models against appropriate baselines and reference benchmarks — and biological interpretation of the features and sequence motifs the model has learned — are essential for both scientific credibility and practical deployment confidence. We provide comprehensive model evaluation and interpretability analysis for every fine-tuning project.
- Comprehensive performance evaluation — AUC-ROC, AUC-PR, F1, MCC, and calibration metric calculation on held-out chromosome test sets; comparison against task-specific state-of-the-art baselines (DeepBind, Basenji2, DeepSEA, SpliceAI, CADD); performance stratification across sequence GC content, repeat content, and variant functional annotation categories; confidence interval estimation by bootstrap resampling
- In silico mutagenesis and variant effect scoring — Systematic single-nucleotide substitution scanning for motif and regulatory feature importance; saturation mutagenesis at key regulatory positions; variant effect score calculation for ClinVar and gnomAD variants; comparison of model variant effect predictions with experimental MPRA and saturation genome editing data
- Attention weight and embedding analysis — Transformer attention head visualisation and motif-attention correlation analysis; JASPAR motif enrichment in high-attention regions; layer-wise attention evolution analysis; UMAP and t-SNE embedding visualisation of genomic sequence representations; sequence similarity clustering from model embeddings
- SHAP and contribution score analysis — SHAP DeepExplainer and GradientExplainer-based nucleotide-level contribution score calculation; contribution score motif enrichment analysis; comparison with ISM (in silico mutagenesis) scores for interpretation consistency; class-specific feature attribution for multiclass genomic classifiers
5. Production Deployment, API Development & Scalable Inference HuggingFace · FastAPI · Docker · AWS · Batch Inference
A fine-tuned genomic foundation model is only valuable if it can be deployed reliably, efficiently, and at scale in a production environment. We provide end-to-end deployment engineering for fine-tuned genomic LLMs — from model quantisation and serving infrastructure through REST API development, containerisation, and integration with existing bioinformatics pipelines.
- Model optimisation for inference — Post-training quantisation (INT8, INT4) with bitsandbytes and AutoGPTQ for reduced memory footprint and faster inference; ONNX export and TensorRT optimisation for GPU-accelerated serving; torch.compile and Flash Attention 2 integration for throughput maximisation; model distillation into smaller task-specific models for edge or embedded deployment
- Serving infrastructure and API development — HuggingFace Text Generation Inference (TGI) and vLLM-based model serving for batched genomic sequence inference; FastAPI REST API development with input validation, async processing, and rate limiting; Docker and Singularity containerisation for reproducible, portable deployment; Kubernetes orchestration for horizontally scalable serving at genomic dataset scale
- Cloud and HPC deployment — AWS SageMaker, GCP Vertex AI, and Azure ML endpoint deployment; spot instance cost optimisation for large-scale batch inference on genomic datasets; HPC SLURM job array configuration for genome-wide variant scoring campaigns; S3/GCS-integrated batch inference pipelines for whole-genome variant effect prediction at population scale
- Pipeline integration and monitoring — Snakemake and Nextflow pipeline integration of fine-tuned model inference steps; model performance drift monitoring in production; input distribution shift detection; prediction confidence monitoring and low-confidence flag alerting; model versioning and rollback capability for production AI governance
Key Applications
Genomic foundation model fine-tuning across regulatory genomics, clinical variant interpretation, protein engineering, and drug discovery.
- Non-coding variant regulatory effect prediction for drug target and GWAS fine-mapping
- Transcription factor binding site prediction in cell-type-specific contexts
- Splice site and splicing regulatory element prediction for clinical variant interpretation
- Promoter and enhancer activity prediction from sequence for regulatory genomics
- Protein function and fitness prediction for directed evolution and protein engineering
- Antimicrobial resistance gene and virulence function prediction from microbial sequences
- Epigenetic mark prediction from DNA sequence for drug target regulatory characterisation
- Gene expression prediction from regulatory sequence for synthetic biology design
Models, Tools & Infrastructure
State-of-the-art genomic foundation models, fine-tuning frameworks, and deployment infrastructure we work with.
- DNA Foundation Models: AlphaGenome, DNABERT-2, Nucleotide Transformer, HyenaDNA, Caduceus, GROVER, GENA-LM
- Regulatory Models: Enformer, Basenji2, DeepSEA, Borzoi, ChromBPNet, Sei
- Cross-Modal Models: Evo, Evo 2, GenSLMs, CodonBERT
- Protein Models: ESM-3, ESM-2, ProtTrans, Ankh, ProGen2, AlphaFold2
- Fine-Tuning: HuggingFace Transformers, PEFT, TRL, bitsandbytes, DeepSpeed, FSDP
- Interpretability: SHAP, Captum, ISM pipelines, Tangermeme, TF-MoDISco-lite
- Deployment: vLLM, TGI, FastAPI, ONNX, TensorRT, Docker, Kubernetes, AWS/GCP/Azure
- Training Infrastructure: PyTorch, Lightning, Weights & Biases, Optuna, SLURM, A100/H100 GPU
- ENCODE / JASPAR / ChIP-Atlas — Reference epigenomic and TF binding datasets for training data curation
- ClinVar / gnomAD / EVE / MAVE — Variant effect and fitness datasets for fine-tuning and benchmarking
Project Deliverables
Structured, production-ready genomic foundation model fine-tuning outputs for every project.
- Model selection report with foundation model comparison and justification
- Training dataset: curated, quality-controlled, and documented sequence data splits
- Fine-tuned model weights and HuggingFace-compatible model card
- Benchmarking report: AUC-ROC, AUC-PR, calibration, and baseline comparison
- In silico mutagenesis and variant effect score files for key genomic regions
- Attention visualisation and SHAP contribution score analysis report
- Inference pipeline: containerised prediction workflow with example usage documentation
- Full written technical report with model design, training, evaluation, and deployment guide
- Production REST API development and cloud endpoint deployment
- Genome-wide variant effect scoring batch inference pipeline
- Model distillation into a smaller, faster task-specific deployment model
- EU AI Act technical documentation and FDA SaMD analytical validation report
- Manuscript methods section and model card for peer-reviewed publication
- Grant application genomic foundation model sections and preliminary results
- Long-term model maintenance, retraining, and performance monitoring retainer
Frequently Asked Questions
Common questions from research groups, pharmaceutical teams, and genomics technology companies.
A genomic foundation model is a large neural network — typically a transformer, state space model, or hybrid architecture — pre-trained on vast quantities of DNA, RNA, or protein sequences using self-supervised objectives such as masked language modelling (predicting masked nucleotides from context) or next-token prediction. This pre-training instils rich, generalisable representations of genomic sequence that capture evolutionary constraints, regulatory grammar, and sequence-function relationships. Fine-tuning then adapts this pre-trained model to a specific downstream task — such as predicting TF binding, classifying pathogenic variants, or scoring splice site strength — by training a task-specific prediction head on a labelled dataset while updating (all or part of) the pre-trained weights. The key advantage is that the model requires far less labelled task-specific data to achieve high performance than training from scratch.
One of the primary advantages of foundation model fine-tuning over training from scratch is substantially reduced labelled data requirements. With parameter-efficient fine-tuning methods such as LoRA, useful task performance can often be achieved with hundreds to a few thousand labelled examples — far less than the tens of thousands required for training a deep learning model from scratch. However, the exact data requirement depends on task difficulty, sequence length, class balance, and the similarity of the task to the foundation model's pre-training objective. For tasks very similar to pre-training (e.g. variant effect prediction from sequence), few-shot performance can be strong; for highly specialised tasks with unusual sequence features, more data is beneficial. We perform data requirement analysis and provide realistic performance projections at project scoping based on your available labelled dataset size.
AlphaGenome (DeepMind, 2025) is designed for genome-wide regulatory prediction — predicting gene expression, chromatin accessibility, TF binding, and variant effects across the full genome at high resolution from long input sequences; it is best suited for regulatory genomics and non-coding variant prioritisation tasks. DNABERT-2 uses a multi-species pre-trained BERT architecture with byte-pair encoding tokenisation — well-suited for general-purpose DNA sequence classification tasks including TF binding, splice sites, and promoter prediction, particularly where labelled data is limited. HyenaDNA uses a state space model architecture enabling context windows up to 1 million base pairs — uniquely suited for tasks requiring ultra-long genomic context such as rare variant modelling and long-range regulatory interaction prediction. We perform a systematic model-task matching analysis at project scoping to identify the most appropriate foundation model for your specific prediction objective.
Yes — and this is one of the most clinically impactful applications. Fine-tuned genomic models can predict the functional consequence of DNA variants on splicing (extending beyond SpliceAI), transcription factor binding disruption, enhancer activity, and chromatin accessibility — providing evidence for ACMG/AMP PP3/BP4 computational prediction criteria in clinical variant classification. However, clinical deployment requires rigorous analytical validation against clinically characterised variant datasets (ClinVar pathogenic variants, MAVE saturation genome editing data), performance benchmarking across variant classes and genomic contexts, and appropriate uncertainty quantification. We design and execute clinically-oriented fine-tuning and validation pipelines aligned with ACMG computational evidence guidelines and, where applicable, EU AI Act and FDA SaMD regulatory requirements.
Absolutely. We assist with the computational genomics and machine learning sections of grant applications — including proposed foundation model selection rationale, fine-tuning methodology, dataset curation approaches, benchmarking design, and preliminary fine-tuning results. We have experience supporting applications to BBSRC, MRC, Wellcome Trust, NIH, and ERC, as well as industry-academic partnership grants. Please contact us as early as possible in the grant preparation process to allow time for any preliminary fine-tuning experiments needed to strengthen the application.
Related Research Areas & Services
Genomic foundation model fine-tuning connects to multiple complementary services we support.
- AI Drug Target Identification — Genomic foundation model variant effect predictions integrated into multi-omics AI target prioritisation; fine-tuned regulatory models for non-coding GWAS variant functional annotation and drug target gene identification
- Clinical Genomics & Variant Interpretation — Fine-tuned genomic models providing PP3/BP4 computational evidence for ACMG/AMP variant classification; splice site and regulatory variant effect prediction for clinical genomics applications
- AlphaFold & Structural Bioinformatics — ESM-3 protein language model fine-tuning for protein engineering complementing AlphaFold2 structural prediction; sequence-structure-function modelling integration
- Epigenomics & DNA Methylation — Enformer and ChromBPNet fine-tuning for epigenomic mark prediction from sequence; regulatory element activity prediction from chromatin accessibility training data
- Clinical AI Validation (EU AI Act) — Regulatory-grade validation of fine-tuned genomic foundation models for clinical deployment; EU AI Act technical documentation and FDA SaMD analytical validation for genomic AI products
- Custom Software & Pipeline Development — Production deployment of fine-tuned genomic foundation models; REST API development, containerisation, genome-wide batch inference pipelines, and model performance monitoring infrastructure
Ready to Fine-Tune a Genomic Foundation Model for Your Research?
Tell us about your genomic prediction task, your available labelled data, your computational environment, and your research or commercial objectives. Our genomic foundation model team will design a tailored fine-tuning and deployment plan — typically within 48 hours of your enquiry. Whether you need AlphaGenome regulatory variant effect prediction, DNABERT-2 sequence classification, HyenaDNA long-context genomic modelling, Enformer expression prediction, Evo cross-modal function prediction, or ESM-3 protein engineering, we are here to deliver expert, validated, and production-ready genomic LLM fine-tuning from day one.
